Disorders Of Calcification
Osteomalacia and Rickets
Clinical Features of Rickets and Osteomalacia
Nutritional Osteomalacia and Rickets
Gastrointestinal and Hepatic Diseases
Impaired Renal Tubular Phosphate Reabsorption
Tumor-Associated Rickets and Osteomalacia
General Renal Tubular Disorders
Hyperparathyroidism, Hypoparathyroidism, and Pseudohypoparathyroidism
Fibrogenesis Imperfecta Ossium
Renal Osteodystrophy and Aluminum Intoxication
Osteomalacia and rickets are disorders of mineralization. Osteomalacia is a failure to mineralize the newly formed organic matrix (osteoid) of bone. In rickets, a disease of children, the growth plate at the epiphysis is involved in a process that is characterized by delay in the maturation of chondrocytes in the growth plate and disorganization of these chondrocytes, resulting in expansion of the growth plate. While rickets is associated with impaired mineralization of cartilage matrix, hypophosphatemia is thought to play a predominant role in the etiology of rickets.1 A number of different disorders are associated with osteomalacia in adults and rickets in children.2–4 The pathogenesis of the mineralization defect, the biochemical alterations, the clinical manifestations, and the therapeutic approaches differ in these conditions, so a systematic approach to the diagnosis and treatment of these disorders is essential.
Mineralization Defect
Mineralization of bone is a complex process in which the calcium-phosphate inorganic mineral phase is deposited in relation to the organic matrix in a highly ordered fashion. Optimal mineralization can take place at bone-forming surfaces only if cellular activity of bone-forming cells is adequate, matrix is normal in composition and is synthesized at a normal rate, the supply of mineral ions (calcium and inorganic phosphate) from the extracellular fluid is sufficient, the pH at sites of mineralization is appropriate (approximately 7.6), and the concentration of inhibitors of calcification is controlled. Structural and regulatory aspects of biological mineralization have been considered in numerous reviews.5–8 Clinical disorders of mineralization can be attributed to defects at several of these control steps, examples of which are shown in Table 15-1. Overall features of mineralization in normal and abnormal bone remodeling and the relevance to mineralization disorders have been comprehensively reviewed.4
Table 15-1
Examples of Disorders with Different Causes of Impaired Mineralization
| Disorder | Mechanism |
| Postoperative hyperparathyroidism | Rate of matrix synthesis exceeds rate of mineralization |
| Fibrogenesis imperfecta ossium | Defective collagenous matrix |
| Adult phosphate diabetes | Phosphate concentration deficient at mineralization sites |
| Vitamin D deficiency | Insufficient calcium and phosphate |
| Systemic acidosis | pH inadequate for mineralization |
| Hypophosphatasia | Excess inhibitor (? inorganic pyrophosphate) |
The determinants for the deposition of the mineral phase in normally mineralized tissues—cartilage, bone, and teeth—and the precise mechanisms that govern the process of biological mineralization have yet to be elucidated. It is thought that deposition of the initial mineral phase takes place within the collagen fibrils in the gap formed by the packing of the trimeric type I collagen molecules, the most abundant organic component of bone.7 The earliest calcium-phosphate (Ca-P) mineral phase is thought to be composed of very small apatite crystals, less than 9 nmol in length, that can fit into the channels formed by adjacent gaps.9 These initial Ca-P crystals appear to form complexes with phosphoproteins of the organic matrix.10 As the bone and minerals mature, additional Ca-P crystals are deposited between collagen macromolecules. Other proteins that are abundant in mineralized tissues and thought to play a role in mineralization include the sialic acid–rich proteins such as osteopontin and bone sialoprotein (BSP).11 The ultimate size and shape of the Ca-P crystals are also influenced by anionic electrolytes in the environment.12
The major driving force for mineralization is the concentration of inorganic phosphate (Pi), which at normal sites of mineralization is derived predominantly from the plasma. Therefore, control of Pi reabsorption in the renal tubular lumen is the most important process regulating mineralization. It is also possible that tissue-nonspecific alkaline phosphatase (TNAP), which functions as an inorganic pyrophosphatase localized on the surface of osteoblasts, generates additional Pi to drive mineralization.13 Osteomalacia is seen in loss-of-function mutations in the TNAP gene, but decreased TNAP also results in accumulation of inorganic pyrophosphate (PPi), which acts as a potent inhibitor of mineralization.13,14 It is likely that this pyrophosphatase function of TNAP is the most critical. PPi is generated by osteoblasts and chondrocytes through the action of two enzymes: the ectonucleoside, pyrophosphatase phosphodiesterase 1 (NNP1/PC-1), which releases PPi from nucleoside triphosphates13,14 and the transmembrane protein, ANK, which shuttles PPi between intracellular and extracellular compartments.15,16 Ectopic mineralization due to decreased local concentrations of PPi accompanies spontaneous deficiency of these proteins in humans and mice. In cartilage, there is evidence that matrix vesicles (MVs) formed from chondrocyte plasma membranes play a role in the regulation of mineralization.5,6 These extracellular MVs are approximately 100 nm in diameter and contain enzymes such as TNAP, NNP1, and Ca-binding proteins as well as Ca-P crystals at the inner surface of the membranes. They are thought to play a role in the initial mineralization of growth plate cartilage matrix by exposing preformed apatite crystals to the extracellular fluid, where further crystal growth can occur. MVs have also been found in bone near sites of mineralization, but it is difficult to assign them a function, since the initial crystallites are deposited in association with collagen fibers in bone. An explanation for the specificity of normal mineralization of bone is provided by the exclusive coexpression in bone of the genes encoding the predominant organic matrix protein, type I collagen, and the enzyme that hydrolyzes the inhibitor, PPi. Furthermore, the protein mineralization inhibitor, matrix GLA protein (MGP), that is expressed in cells in several other tissues is not expressed in bone.17,18
The mechanism of defective mineralization is not the same in all disorders associated with osteomalacia and rickets, and biochemical indices such as serum levels of calcium and phosphorus also differ. Moreover, the relative imbalance in matrix synthesis and its mineralization varies depending on the underlying disease mechanism. Estimates from tetracycline labeling indicate that the appositional growth rate in normal bone is about 1 µm/day.19 It has also been suggested20 that complete mineralization of the osteoid in normal bone requires approximately 10 to 21 days. Thus the thickness of the osteoid seam normally does not exceed 15 to 20 µm, the surface of bone that is covered by osteoid is normally less than 20%, and the active surface that is covered by osteoid is considerably less. The major histologic criteria for the diagnosis of osteomalacia are the increased osteoid surface and the increased thickness of the osteoid seam (Fig. 15-1). The mineralization front at the junction of mineralized bone and osteoid is also abnormal in osteomalacia.21 In applying kinetic criteria to the diagnosis of osteomalacia, it has been suggested that a mean osteoid seam width greater than 15 µm and a mineralization lag time greater than 100 days are appropriate diagnostic criteria.22 However, other investigators23,24 have suggested that more stringent criteria be applied to establish the diagnosis of osteomalacia, based on the observation that reduced mineral apposition rate, reduced fractional extent of the mineralization front, and prolongation of the mineralization lag time are indices that reflect impaired matrix synthesis by osteoblasts rather than specific features of osteomalacia. The diagnosis of osteomalacia must include evidence of an absolute increase in the total osteoid volume and an increased number of osteoid lamellae. A detailed consideration of the kinetics of remodeling as specifically applicable to osteomalacia is found in a review by Parfitt.4

FIGURE 15-1 Undecalcified sections of a bone biopsy from a patient with an adult-onset renal phosphate leak. A, Unstained. B, Microradiograph. C, Ultraviolet photomicrograph demonstrating fluorescence (F) of tetracycline administered 14 days prior to biopsy. Mineralized bone (M) and osteoid (O) are noted. (From Case 1 in Baylink D, Stauffer M, Wergedal J et al: Formation, mineralization, and resorption of bone in vitamin D–deficient rats, J Clin Invest 49:1122–1134, 1970.)
The architecture of the bone cells and matrix in osteomalacic bone is usually normal. The collagen of the osteoid is largely lamellar, although foci of woven bone are occasionally seen. Hypomineralized periosteocytic lesions have been observed in some affected individuals with hypophosphatemic rickets.25 The persistence of this defect in patients in whom the abnormality in bone mineralization was corrected with therapy supports the hypothesis that osteocyte function may be abnormal. In contrast, there are clear abnormalities in the cells of the rachitic growth plate. The characteristic changes occur in the maturation zone of hypertrophic chondrocytes, whereas the resting and proliferative zones show normal histologic features (Fig. 15-2). In the maturation zone, the number of cells per column is increased, and the cells are irregularly aligned. This is also accompanied by an increase in the transverse diameter, which may extend beyond the ends of the bone, resulting in characteristic cupping or flaring. In experimental rickets, the water content of the growth plate is increased, and a number of metabolic abnormalities have been observed, including decreased glycogen content and an altered pattern of glycolysis.26 When bone is examined histologically, it is essential that undemineralized sections be used. In usual practice, however, with classic clinical, radiologic, and biochemical findings, bone biopsy is not necessary to arrive at the diagnosis of osteomalacia. The most commonly biopsied site is the iliac crest; sample size ranges from 5 to 10 mm in diameter and should include both inner and outer cortices. Growth plates from long bones in children are usually not biopsied, although an open-wedge biopsy of growth cartilage of the iliac apophysis may occasionally be obtained without the hazard of altering growth of long bones. Mineralized specimens of bone are most satisfactorily embedded in plastic media—which provide preservation of tissue architecture not usually attained with paraffin-embedding techniques—because the distinction between mineralized and unmineralized bone is lost with decalcification of the specimen. A number of different staining techniques can then be used to demonstrate the osteoid and apply quantitative morphometric analysis.23–25 In normal bone, the mineralization front is seen at the junction of the osteoid seam and newly mineralized bone. This region can be identified by an intense fluorescence of tetracycline, deposited in this zone when administered prior to obtaining the biopsy (see Fig. 15-1). In normal persons, the osteoid seam/bone junctions fluoresce intensely; in osteomalacia, the fluorescence is less well defined (more diffuse) or even absent. In addition to impaired matrix mineralization, matrix biosynthesis may be abnormal in osteomalacia. In osteomalacia observed with vitamin D deficiency, a decreased rate of matrix formation is observed.23,24,27 Osteoblast function may be impaired in many forms of human rickets and osteomalacia, which may result in abnormal matrix formation. Notably, the hydroxylation of certain collagen lysyl residues is increased in vitamin D–deficient bone, as well as in other experimental hypocalcemic states.28–30

Clinical Features of Rickets and Osteomalacia
The clinical manifestations of rickets are mainly related to skeletal pain and deformity, slippage of epiphyses, disturbances in growth, and fracture of the osteomalacic bones. Hypocalcemia, when it occurs, may be symptomatic. Depending on the degree of hypophosphatemia, muscular weakness and hypotonia may be prominent features. Dent and Stamp31 have indicated nine factors that underlie the clinical manifestations of rickets and osteomalacia, modified here as follows:
1. Failure of mineralization affects predominantly those parts of the skeleton in which growth is most rapid.
2. Endochondral bone is more affected than intramembranous bone, possibly because of the more rapid growth of the former.
3. Proximal and distal ends of bones do not grow at the same rate, and rickets affects the most rapidly growing area.
4. Because different bones grow at different rates at different stages of development, the time when rickets is active determines the clinical expression. For example, the skull is growing rapidly at birth, so craniotabes is a manifestation of congenital rickets. During the first year, the upper limbs and rib cage grow rapidly, so abnormalities at these sites are prominent—for example, rachitic rosary. Signs of rickets at the wrist are usually seen at the ulnar side because the growth rate of the distal ulnar epiphysis is greater than that of the distal radial epiphysis.
5. Deformities in mild chronic rickets are most often due to disordered growth at the epiphysial plate rather than to bending at the shafts.
6. In some forms of rickets, the radiologic changes include those of secondary hyperparathyroidism (subperiosteal resorption, most commonly at the metaphyses).
7. Deformities that occur before the age of 4 years correct themselves if the rickets is cured; if rickets persists to a later age, the deformities are permanent (dwarfism, bowleg, and knock-knee).
8. “Late” rickets, which occurs at the time of the pubescent growth spurt, produces dramatic disturbances and results in knock-knee.
9. Adult manifestations of osteomalacia, such as Looser’s zones and increased biconcavity of vertebral bodies, are seen in young children only when the rickets is very severe.
In contrast, osteomalacia in adults may be difficult to detect on clinical grounds alone. Diffuse skeletal pain and muscular weakness may be present. Pain, often prominent about the hips and in association with hypophosphatemic myopathy, may produce a waddling or antalgic gait. Fractures may occur in the ribs and vertebral bodies, as well as in long bones, leading to progressive deformities. Affected individuals may also have localized pain and swelling in one or more joints. Synovial fluid is noninflammatory and free of crystals. Symmetric polyarthralgias resembling those of rheumatoid arthritis or polymyalgia rheumatica may also be observed.32 Muscular weakness is quite common,33,34 is primarily proximal in distribution (which contributes to the waddling gait), and is often associated with wasting and hypotonia with preservation of brisk reflexes.35 This is thought to be a consequence of hypophosphatemia and responds to phosphate repletion.36 The molecular basis remains elusive, with no difference observed in the relative concentrations of skeletal muscle phosphocreatine, adenosine triphosphate, or inorganic phosphate estimated by phosphorus nuclear magnetic resonance spectroscopy.37 Although the etiology of the neuromuscular features of osteomalacia is not clearly defined, therapy of the underlying disorder, such as vitamin D in nutritional osteomalacia, alkalinization in acidosis, and phosphate repletion in hypophosphatemic osteomalacia, results in resolution of these features. The role of hypophosphatemia per se in muscular weakness is discussed in Chapter 6.
Radiologic Features
Radiologic features of rickets and osteomalacia reflect the histopathologic changes. In rickets, the alterations are most evident at the epiphyseal growth plate, which is increased in thickness, cupped, and reveals haziness at the diaphyseal border due to decreased mineralization of the hypertrophic zone and inadequate mineralization of the primary spongiosa (Fig. 15-3). Variation in the pattern of rachitic changes is influenced by differences in the rates of growth of individual bones. The trabecular pattern of the metaphyses is abnormal, the cortices of the diaphyses may be thinned, and bowing of the shafts may be present.
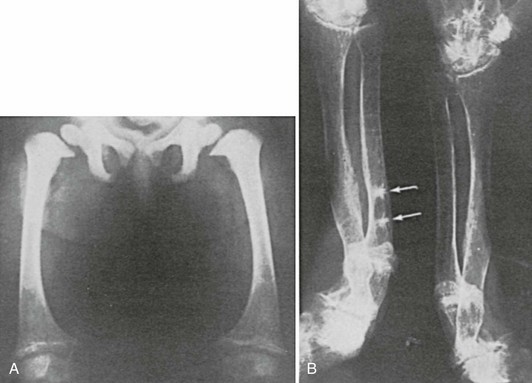
FIGURE 15-3 A, Rickets in a child with Fanconi’s syndrome, showing typical cupping of distal femoral epiphyses. B, Osteomalacia in an 80-year-old woman who had a history compatible with hypophosphatemic rickets dating to early childhood. Note multiple pseudofractures (arrows).
Osteomalacia is due to decreased mineralization and is therefore associated with a decrease in bone density, loss of trabecular patterning, and variable degrees of thinning of the cortices.38,39 In some patients, radiologic changes are indistinguishable from those seen in osteoporosis. The characteristic finding that specifically suggests osteomalacia is the presence of radiolucent bands known as pseudofractures, Looser’s zones, or umbauzonen, ranging from a few millimeters to several centimeters in length and usually oriented perpendicularly to the surface of the bone. (Fig. 15-4). They tend to occur symmetrically and are particularly common at the inner aspects of the femur near the femoral neck, in the pelvis, in the outer edge of the scapula, in the upper fibula, and in the metatarsals.
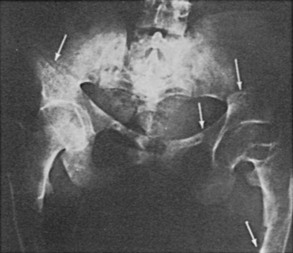
FIGURE 15-4 Radiograph of the pelvis and proximal femora in an adult with renal phosphate wasting. Note pseudofractures, also known as Looser’s zones (arrows). (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
Pseudofractures are most often seen at sites where major arteries cross the bones. Arteriography in some36,40,41 but not all cases42 suggests that the origins of the pseudofractures correspond to the locations of major arteries (Fig. 15-5). Trauma of some sort, whether related to arterial pulsation or other factors (e.g., weight-bearing stress), is likely responsible for the symmetry of the lesions and their predilection for the described sites. Pseudofractures are often multiple, occasionally occurring at 10 to 15 sites in a single individual; such multiple symmetric pseudofractures in osteomalacic individuals have been referred to as Milkman’s syndrome.43–46 The abnormalities in Milkman’s original case were also considered by Albright and Reifenstein43 to be manifestations of osteomalacia. The histopathology of Looser’s zones is that of premalacic lamellar bone, some of which is surrounded by lamellar osteoid at the edge of the defect.47 In addition, there are foci of woven bone, some of which is mineralized and some not. This accounts for the lower radiologic mineral density of the pseudofracture compared with the surrounding bone. Subperiosteal erosions along the diaphyseal cortices extending to the metaphyses may be seen when secondary hyperparathyroidism is present. Widening (or pseudowidening) of the sacroiliac joints and the appearance of hazy margins has also been observed, sometimes suggesting ankylosing spondylitis, which osteomalacia may mimic clinically.36
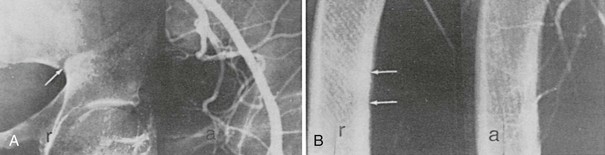
FIGURE 15-5 Radiograph (r) and corresponding arteriograms (a) of a patient with adult-onset renal phosphate wasting. A, Pelvis. B, Femur. Note that the origin of Looser’s zones (arrows) corresponds with crossing of major vessels. (From Case 2 in Jaworski ZFG, Kloswvych S, Cameron E: Proceedings of the First Workshop on Bone Morphometry. Ottawa: University of Ottawa Press, 1973.)
In some patients with osteomalacia, increased rather than decreased radiologic density of bones may be observed.48 This is seen particularly in patients with renal tubular phosphate leaks, as opposed to vitamin D deficiency (Fig. 15-6). In such patients, there may be a striking degree of thickening of the cortices and trabeculae of the spongy bone, at times associated with exostotic spurs. This hyperostosis has been noted in untreated patients. It is not usually observed in patients with generalized defects in proximal renal tubular reabsorption. Despite the increase in mass of bone per unit volume, microscopically the trabeculae are covered with abnormally thickened osteoid seams typical of osteomalacia. Similar findings may be noted in patients with chronic renal failure. The reason for the hyperostosis is unknown; the bone is still architecturally abnormal and subject to fracture with relatively minimal trauma.
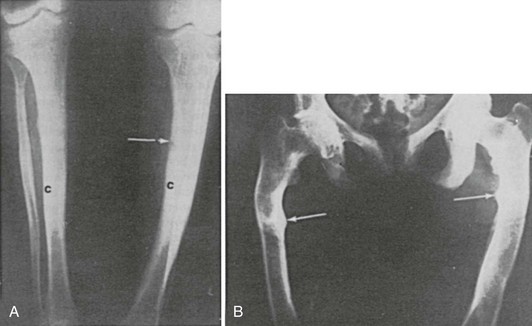
FIGURE 15-6 Increased bone mass in patients with osteomalacia. A, Radiographs of femora of a 15-year-old boy with X-linked hypophosphatemia. Note the thick tibial cortex (c) and Looser’s zone (arrow). B, Radiograph of pelvis and femora of a 38-year-old woman with hypophosphatemia present since childhood. Note Looser’s zones (arrows).
In patients with X-linked hypophosphatemic osteomalacia and rickets, an additional finding has been the presence of a generalized involvement of the entheses, with exuberant calcification (more likely ossification) of tendon and ligament insertions.49,50 The absence of inflammatory cells, as well as other clinical features, differentiates this disorder from degenerative joint disease and the seronegative spondyloarthropathies. A comprehensive classification of rickets and osteomalacia is shown in Table 15-2. A detailed discussion of all these conditions is not included here.
Table 15-2
Classification of Rickets and Osteomalacia
B Impaired renal tubular phosphate reabsorption (intestinal)
VII General renal tubular disorders (Fanconi’s syndrome)
VIII Primary mineralization defects
IX States of rapid bone formation with or without a relative defect in bone resorption
Nutritional Osteomalacia and Rickets
In 17th century Scotland and England, the association of poverty and malnutrition with the occurrence of infantile rickets was vividly documented. The widespread prevalence of infantile rickets in the industrialized regions of Britain was further documented in reports from Glasgow in the late 1800s and early 1900s. The link between rickets, dietary deficiency of vitamin D, and correction of vitamin D deficiency by solar radiation was finally established in 1923 by the work of the Vienna Council. Following this discovery, fortification of certain foods with vitamin D reduced the incidence of nutritional rickets in Europe and the United States to negligible levels, and by the 1940s, vitamin D deficiency was no longer regarded as an important cause of osteomalacia and rickets.43 Vitamin D metabolism and the role of specific metabolites in bone development, mineralization, and remodeling are discussed elsewhere (see Chapter 3). Nevertheless, the role of vitamin D in the prevention of osteomalacia and rickets is relevant to the discussion in this chapter.
Studies in Glasgow51–53 and London54 documented the reappearance of nutritional osteomalacia and rickets as a public health problem in the United Kingdom. Since the 1950s, the population at risk primarily involves East Asian immigrants whose unique dietary and social customs have led to the development of osteomalacia and rickets.52,54–58 Other groups at risk include housebound and elderly subjects and food faddists, especially those on vegetarian or fat-free diets.52,54–58 An unexpectedly high prevalence of osteomalacia was also observed in elderly women with rheumatoid arthritis who were housebound and had poor nutritional status.59
Gastrointestinal and Hepatic Diseases
Another population at risk for development of nutritional osteomalacia are morbidly obese individuals60 and those who have undergone intestinal bypass surgery for treatment of severe obesity.61–63 In one bypass surgery series, iliac bone biopsies revealed the presence of osteomalacia in nearly a third of 21 patients studied.63 Treatment with vitamin D2 (36,000 IU/day), as well as supplemental calcium (27 mmol/day), was required to promote a more positive calcium balance in some individuals.61 An increased frequency of osteomalacia also was observed in patients after gastrectomy,64–66 the severity of the mineralization defect being positively correlated with serum 25-hydroxyvitamin D [25(OH)D] but not with serum 1,25-dihydroxyvitamin D [1,25(OH)2D].
Rickets occurs in infants and children with cholestatic liver disease.67 Children with biliary atresia develop biliary cirrhosis, jaundice, and ascites. Intestinal absorption of vitamin D is markedly impaired, and serum values are low. Bone disease has been attributed to vitamin D deficiency, and pharmacologic doses of 1,25(OH)2D3 are required for treatment of the bone disease.68
Low-birthweight infants are at risk for rickets, with the reported incidence ranging from 13% to 32%. Insufficient intake of calcium, phosphorus, and vitamin D have been implicated. The condition should be detected early, since prompt nutritional supplementation is required.69 Infants of vitamin D–deficient mothers are at greatest risk for neonatal rickets,70 which can be averted by maternal vitamin D supplementation during pregnancy. Special consideration should be given to screening for vitamin D deficiency during pregnancy in women whose social and dietary history reveal inadequate sun exposure and food sources of vitamin D. Infants who are entirely breastfed and have inadequate exposure to sunlight are also at risk for rickets, because the amount of vitamin D and 25(OH)D in human milk is inadequate.71
Vitamin D sources include dietary supplementation with ergocalciferol (vitamin D2), an irradiation product obtained from plants, and cholecalciferol (vitamin D3), produced in human skin by the action of ultraviolet light on the physiologic precursor, 7-dehydrocholesterol. Because most foods (with the exception of fatty fish) contain only small amounts of vitamin D3, individuals must rely on either adequate sunlight exposure or dietary supplements to avoid vitamin D deficiency. There is marked seasonal variation in plasma 25(OH)D, independent of age and sex.72–74 These variations parallel changes in duration and intensity of sun exposure, with higher values in late summer months in the northern hemisphere. Plasma 25(OH)D is almost twice as high in American women of Caucasian descent than in those of African descent during winter months, and the increment during summer months is also greater.75 Plasma 25(OH)D and parathyroid hormone (PTH) are inversely correlated, implying that the changes in circulating 25(OH)D are metabolically significant.76 In a study of patients who were hospitalized in a general medical ward, plasma 25(OH)D was low in over half of the subjects, consistent with a high prevalence of vitamin D deficiency.77 Thus, adequate vitamin D supplementation is essential when exposure to sunlight is marginal.
Vitamin D requirements are greater in the elderly than in young adults. This difference is attributed to an age-related decline in dermal production of 7-dehydrocholesterol, the precursor for vitamin D3,78 diminished renal production of 1,25(OH)2D,79 and diminished intestinal absorption of calcium caused by lower levels of the vitamin D receptor in the intestine.80 Other contributing factors include previous gastric surgery and occult malabsorption in addition to altered vitamin D metabolism. To determine optimal plasma 25(OH)D and how much vitamin D is required to achieve optimal values, Vieth,81 in an exhaustive review, and Heaney in another82 concluded that total daily intake and production of vitamin D of 2.5 to 5.0 mg (100 to 200 IU) and plasma 25(OH)D values greater than 20 to 25 nmol/L are sufficient to prevent clinical rickets or osteomalacia. Higher intakes or production of vitamin D would be necessary, however, to prevent secondary hyperparathyroidism, bone loss, and subclinical osteomalacia. Therefore, it may be necessary to maintain serum 25(OH)D levels of 100 nmol/L or greater to avoid bone loss, subclinical osteomalacia, and osteoporosis. The amount of vitamin D supplementation necessary to attain these levels varies widely, based on dietary factors and solar exposure.
In the East Asian immigrant populations of Britain, rickets and osteomalacia secondary to vitamin D deficiency are seen most commonly in neonates, infants, and adolescents during pubertal growth and less frequently among adults.51,83–85 Multiple factors have been implicated in the development of bone disease, including insufficient intake of calcium and vitamin D, skin pigmentation,86 which attenuates ultraviolet transmission through the epidermis, genetic factors,87–89 and social customs, such as avoidance of sun exposure and consumption of chapati, a dietary staple flatbread high in phytate, which binds calcium in the gut and interferes with its absorption.90 Furthermore, studies in rats demonstrated that the rate of inactivation of vitamin D in the liver was increased by a calcium-restricted diet.
Studies by Dent and colleagues91 strongly suggest that insufficient sunlight exposure plays a pivotal role in the development of nutritional rickets and osteomalacia, in addition to the rickets and osteomalacia observed in patients on long-term anticonvulsant therapy.92 In two carefully studied individuals, they demonstrated healing of rickets and positive calcium balance following therapy with ultraviolet light, despite a vitamin D–deficient, high-phytate diet. Substitution of a low-phytate diet did not affect the plasma biochemical abnormalities or the calcium balance.91
Scriver93 divides the evolution of vitamin D deficiency in infancy into three stages. In stage 1, serum calcium tends to be low, serum phosphorus normal, and aminoaciduria absent. Without treatment, stage 2 develops, and aminoaciduria and hypophosphatemia appear as a consequence of diminished tubular reabsorption. In this stage, serum calcium tends to return to normal, and serum alkaline phosphatase increases, presumably related to the increased PTH and resultant increase in bone turnover. Renal tubular dysfunction (aminoaciduria, phosphaturia) has, at least in part, been attributed to the increased PTH.94,95 Stage 3 is characterized by the return of hypocalcemia. The effect of lowered concentrations of serum phosphorus in stage 2 and lowered concentrations of serum calcium and serum phosphorus in stage 3 presumably account for development of the mineralization defect.
Rao et al.96 reviewed the histomorphometric findings in a series of 65 patients with vitamin D depletion diagnosed on the basis of plasma levels of 25(OH)D less than 10 ng/mL. They found that in early vitamin D depletion, the effects on bone are manifested principally by the occurrence of secondary hyperparathyroidism. With increasing severity or duration of the vitamin D deficiency, the mineralization process becomes progressively impaired, bone formation rates decline, and osteoid surface and thickness increase.
In summary, rickets and osteomalacia are being recognized with increasing frequency in selected populations. Serum 1,25(OH)2D is not a reliable indicator of vitamin D nutrition, since values may be normal in individuals with vitamin D deficiency.97,98 The availability of serum 25(OH)D assays has also permitted detection of those at risk, prior to the development of overt clinical disease.
Acidosis and Osteomalacia
Acidosis resulting from a number of different causes has been associated with osteomalacia. The mechanism of bone loss and the mineralization defects are complex and not completely understood. Albright and Reifenstein43 originally suggested that acidosis produces slow dissolution of the mineral phase of bone in an attempt to buffer retained hydrogen ion. This process is associated with hypercalciuria. Support for this suggestion has been obtained in studies of patients with renal tubular acidosis in whom retention of hydrogen ion is greater than that theoretically required to produce the observed decrease in plasma bicarbonate concentrations.99 On the basis of measurements in normal subjects in whom metabolic acidosis is induced, it has been proposed that excess retained hydrogen ion is balanced by bone buffering and loss of bone calcium in the urine.100–102 Since the hypercalciuria and increased bone resorption that accompany most acidotic states do not directly result in osteomalacia, other mechanisms must be invoked to explain the occurrence of clinically significant skeletal mineralization defects. Osteoclasts function optimally at a pH of approximately 6.9 and are inactive at a pH greater than 7.3; therefore, it is probable that the calcium release from bone induced by acidosis can be ascribed to increased osteoclast activity rather than to simple physicochemical buffering.103 Chronic acidosis activates vacuolar hydrogen ion pumps in isolated osteoclasts and stimulates osteoclastic bone resorption.104 In addition, activation of the voltage-gated H+ channel through protein kinase C in osteoclasts can sense changes in pH.105
In parallel, metabolic acidosis inhibits the function of osteoblasts: in osteoblast culture systems, lowering pH to 6.9 decreases formation of mineralized nodules.106 Acidosis increases expression of osteoblastic RANK-L via a cyclooxygenase-dependent mechanism, leading to enhanced osteoclastogenesis.107 Another potential proton-sensing mechanism in the osteoblast is G protein–coupled receptors OGR1 and OGR2.108 OGR1 is inactive at pH 7.8 and fully activated at pH 6.8, signals through the phosphoinositol pathway, and is expressed in osteosarcoma lines and primary osteoblasts. OGR2 signals through the cyclic adenosine monophosphate (cAMP) pathway but has not yet been shown to be expressed in bone cells. Maintenance of pH within a critical range is thus essential for bone-cell function and for mineralization to proceed normally.
Acidosis can also affect phosphate metabolism by altering renal tubular handling of the anion. In patients with chronic acidosis, treatment with alkali to correct the acidosis can normalize serum phosphate by increasing renal tubular phosphate reabsorption and phosphate maximal tubular excretory capacity.109,110 Secondary hyperparathyroidism may be another factor involved in the altered phosphate handling in systemic acidosis,109 and acidosis can impair intestinal calcium absorption in response to exogenous vitamin D,106 as well as activation of 25(OH)D.111 Rickets and osteomalacia secondary to acidosis are most frequently a complication of inherited distal renal tubular acidosis (RTA).112 Clinical manifestations vary widely in type as well as severity. Autosomal-dominant as well as autosomal-recessive forms have been described. As a rule, the dominant form is usually recognized in adults who present with relatively mild disease characterized by nephrolithiasis and acidosis. Osteomalacia occurs infrequently. In contrast, the autosomal-recessive form is recognized in infancy or early childhood, is usually severe, and rickets is common. In dominant RTA, the causal mutation is in the SLC4A1:AE1 gene that encodes the heterotopic Cl−/HCO3− exchanger located in the α-intercalated cells of the distal tubules. In recessive RTA, the causal mutations are in the B1 or A4 subunits of the H+/ATPase encoded by ATP6V1B1 and ATP6V0A4 genes, respectively. Rarely, mutations in the SLC4A1:AE1 gene occur in recessive RTA as well. In most of the cases described in early reports,113–115 healing of the bone disease resulted from correction of the acidosis with sodium bicarbonate alone (5 to 10 g/day). Nevertheless, healing is slow, and the response may be hastened by the addition of vitamin D or 1,25(OH)2D3. Occasionally, vitamin D toxicity can develop unexpectedly, so patients must be monitored carefully. Although chronic treatment with vitamin D is not necessary once the osteomalacia is cured, continued use of vitamin D may be required to complete healing in those individuals in whom the glomerular filtration rate is low.115,116
Another form of inherited mixed distal/proximal RTA is associated with osteopetrosis and cerebral calcifications. The osteopetrosis, due to inadequate osteoclast function secondary to defects in acidification in the extracellular space adjacent to the ruffled border, is caused by mutations in the gene that encodes carbonic anhydrase II (CAII).117–119 This RTA is characterized by defective urinary acidification and bicarbonate wasting, and the syndrome is explained by the fact that CAII is expressed not only in osteoclasts but in both proximal and distal segments of the nephron. It is of interest that bone marrow transplantation does not affect the acidosis but reverses the osteopetrosis (by radiologic and histologic criteria), since osteoclasts are of hematopoietic origin.120
In several of the syndromes associated with more widespread renal tubular reabsorptive defects, systemic acidosis may contribute to the pathogenesis of osteomalacia. Some of these are inherited, such as various forms of Fanconi’s syndrome and Lowe’s syndrome (oculocerebrorenal syndrome). Some patients with renal tubular phosphate leaks may also have mild acidosis.121–125 Other aspects of these syndromes are considered elsewhere in this chapter.
Related posts:
 Hereditary Disorders of The Skeleton
Hereditary Disorders of The Skeleton
 Calcitonin
Calcitonin
 Bone Development and Remodeling
Bone Development and Remodeling
 Malignancy-Associated Hypercalcemia and Medical Management
Malignancy-Associated Hypercalcemia and Medical Management
 Parathyroid Hormone and Parathyroid Hormone–Related Peptide In The Regulation of Calcium Homeostasis and Bone Development
Parathyroid Hormone and Parathyroid Hormone–Related Peptide In The Regulation of Calcium Homeostasis and Bone Development
 Bone Density and Imaging of Osteoporosis
Bone Density and Imaging of Osteoporosis
