Cancer of the Endocrine System
David F. Schneider, Haggi Mazeh, Sam J. Lubner, Juan C. Jaume and Herbert Chen
• The incidence of thyroid cancer is increasing, and there are approximately 33,500 new cases per year in the United States.
• The incidence of differentiated thyroid cancer is 11.6 per 100,000 people per year, with a female-to-male ratio of more than 3 : 1.
• Differentiated thyroid cancer (DTC) includes papillary thyroid cancer (PTC), which accounts for 80% of all thyroid cancers, follicular thyroid cancer (FTC), which accounts for 10% to 20% of all thyroid cancers, and a rare type, Hürthle cell cancer.
• Medullary thyroid cancer (MTC) arises from the parafollicular C cells and accounts for 5% to 10% of all thyroid cancers.
• Anaplastic thyroid cancer is a rare, but rapidly fatal, form of thyroid cancer.
• Other histologic types of cancer, such as lymphoma, sarcoma, and metastatic cancers, can also be found within the thyroid.
• Known risk factors for the development of thyroid cancer include radiation exposure and iodine deficiency.
• Thyroid cancer can also run in families or exist as part of familial syndromes (Gardner, Cowden, and Werner syndromes)
• More recently, the molecular pathogenesis of thyroid cancer has been investigated. The following are the most widely studied molecular markers for DTC to date:



• Well-differentiated histology has excellent 5-year survival (>95%).
• Older age and extent of invasion are related to prognosis.
• Lymph node involvement is associated with higher recurrence, but has questionable impact on survival.
• Many staging systems exist for DTC.
• Hürthle cell adenoma—larger size (>6 cm) predicts malignancy.
• Poorly differentiated tumors—recurrence lymph node metastases are common.
• Anaplastic cancers are extremely aggressive with 5-year survival less than 5%.
• The history should include radiation exposure, family history, and compressive symptoms (dysphagia, hoarseness, pain/pressure) from enlarging tumor.
• Concerning exam findings, include a fixed mass or lymphadenopathy.
• Incidental fining on computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET), and ultrasound.
• Preoperative laboratory studies include thyroid-stimulating hormone (TSH), Tg.
• Fine-needle aspiration (FNA) biopsy is a key component of the workup of thyroid nodules. The Bethesda Criteria classify FNA results and determine the risk of cancer in the nodule.
• Preoperative imaging should cervical ultrasound. CT is used when aggressive variants are suspected to assist in operative planning.
• Treatment begins with surgery. Most thyroid cancers are treated with total thyroidectomy. Compartment-oriented neck dissection is added when there is metastatic disease in the cervical lymph nodes.
• Adjuvant therapy for differentiated tumors is radioactive iodine (131I).
• Patients must be prepared for radioactive iodine ablation with a low iodine diet.
• And thyroid hormone withdrawal or recombinant human thyroid-stimulating hormone (rhTSH) if there is no evidence of metastatic disease.
• After surgery and radioactive iodine, thyroxine-suppression prevents the growth of microscopic disease.
• External beam radiation is utilized for persistent, recurrent, anaplastic, poorly differentiated tumors that are not iodine avid.
• Chemotherapy is mainly palliative for poorly differentiated or anaplastic tumors. Traditional chemotherapy has minimal response rates, but newer, targeted therapies, such as sorafenib or sunitinib, are showing promise.
• Surveillance for recurrent thyroid cancer includes measurements of TSH, Tg, and antithyroglobulin antibodies in addition to cervical ultrasound. The schedule of these tests is tailored to risk level.
• Treatment of recurrence can include external beam radiation or targeted therapies, depending on the iodine avidity of the tumor.
• Medullary thyroid cancer accounts for 5% to 10% of all thyroid cancers; 75% of cases are sporadic and 25% are familial (multiple endocrine neoplasia [MEN]-2, familial medullary thyroid carcinoma [FMTC]).
• The diagnosis is made by FNA with calcitonin washout. RET testing can identify inherited germline mutations. As in DTC, cervical ultrasound assists with operative planning. The tumor markers calcitonin and carcinoembryonic antigen (CEA) can be useful in following patients postoperatively for identifying recurrence and metastases.
• At a minimum, treatment of MTC should consist of total thyroidectomy plus central lymph node dissection. Lateral neck dissection is added when there are clinically positive nodes in the central neck and for high-risk patients.
• Traditional chemotherapy is not effective for metastatic MTC, but newer, targeted therapies for metastatic disease such as vandetanib, have shown some promise.
• The incidence of adrenocortical cancer is 1 to 2 per million people.
• Most adrenocortical cancers are sporadic, but they can also occur as part of familial syndromes such as MEN-1, Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome, and Carney complex.
• Most are asymptomatic, but 40% to 60% are functional (hormone production), and this may be the presenting symptom(s).
• The diagnosis is by urinary/plasma biochemical testing and imaging: CT, fluorodeoxyglucose (FDG)-PET.
• Often, the diagnosis is not made definitively until after resection of suspicious masses, and pathology provides definitive diagnosis. FNA of adrenal masses is rarely indicated.
• Surgery is the mainstay of treatment for adrenocortical cancer and should consist of en bloc resection of the adrenal gland with adjacent organs/tissue that is involved; cardiopulmonary bypass may be necessary for caval involvement.
• Long-term surveillance, consisting of physical exams and CT scans, is necessary to monitor for disease recurrence.
• Mitotane alone or in combination with other chemotherapeutic agents improves recurrence-free survival.
• Radiotherapy may improve local control, but there are no clear recommendations.
• Hormonal control can also limit disease spread and consists of mitotane, ketoconazole, metyrapone, etomidate.
• Excision or reoperation is recommended for recurrent or metastatic disease.
• The prognosis is poor, with overall 5-year survival of less than 40%.
• The incidence of malignant pheochromocytoma is 2 to 8 per 1,000,000 adults.
• Most malignant pheochromocytomas are sporadic, but 10% are part of inherited syndromes such as MEN syndromes, neurofibromatosis type I, von Hippel-Lindau syndrome, and succinate dehydrogenase gene mutations.
• Pheochromocytomas present with the classic triad of functional tumors-headache, tachycardia, and sweating, but they are asymptomatic in more than 50%, presenting as an incidental adrenal mass.
• The diagnosis is established with urinary or plasma fractionated metanephrines and catecholamines.
• Tumors are localized with CT/MRI; metaiodobenzylguanidine (MIBG) to identify extraadrenal metastases.
• Treatment begins with surgery to resect entire gland with clear margins. Surgery can also be useful in debulking for metastatic disease.
• Metastatic or unresectable disease can be treated with 131I-MIBG or chemotherapy.
• Radiotherapy is used for palliation in bone and lymph node metastases.
• Medical therapy to prepare patients for surgery or control symptoms of catecholamine excess can include phenoxybenzamine, nicardipine, or metyrosine.
• The 5-year survival ranges from 20% to 50%.
• MEN-1 presents first with hyperparathyroidism in their 30s and 40s. Other manifestations include pituitary tumors and neuroendocrine tumors of the pancreas (such as gastrinoma) and upper gastrointestinal tract.
• MEN-1 is inherited in an autosomal-dominant fashion. Mutations in the MENIN tumor suppressor gene cause this disease, but expression is variable.
• Treatment of parathyroid hyperplasia is subtotal parathyroidectomy and bilateral cervical thymectomy.
• MEN-1 patients with pancreatic and duodenal tumors are treated with distal pancreatectomy, enucleation of pancreatic head tumors, duodenotomy, and mucosal resection of multiple duodenal tumors.
• MEN-2A is characterized by pheochromocytoma, MTC, and primary hyperparathyroidism (hyperplasia).
• MEN-2B is characterized by more aggressive MTC, pheochromocytoma, and mucosal ganglioneuromas.
• The MTC in MEN-2 syndromes arises from C-cell hyperplasia and germline RET mutations.
• Specific codon mutations in the RET gene determine the disease phenotype in MEN-2 syndromes, and help risk-stratify patients.
• Prophylactic thyroidectomy should be offered to mutation carriers; the timing of thyroidectomy is determined by the specific codon mutation.
• Pheochromocytomas are often bilateral in MEN-2 syndromes, but onset is asynchronous. Consequently, prophylactic adrenalectomy is not indicated. Those MEN-2 patients who develop bilateral disease can be treated with bilateral adrenalectomy and hormone replacement or cortical-sparing adrenalectomy.
• The incidence of carcinoid tumors is 5.25 in 100,000 people.
• Tumors are identified with specific immunohistochemical staining for NSE or chromogranin A. Chromogranin A also serves as a blood marker for the disease.
• Several classification systems exist for carcinoid tumors, including the World Health Organization (WHO) classification and European Neuroendocrine Tumor Society (ENETS) staging system.
• Carcinoids arise from the Kulchitsky cells in crypts of Lieberkühn of the gut or disseminated in the endobronchial mucosa, and are classified by location.
• The diagnosis is made definitively by tissue diagnosis, but urinary 5-hydroxyindoleacetic acid (5-HIAA), serum NSE, and chromogranin A are serum markers of the disease.
• CT or MRI can localize the carcinoid tumors, or OctreoScan can be used as these tumors have somatostatin receptors.
• The carcinoid syndrome occurs in metastatic carcinoid and presents as flushing, diarrhea, and bronchoconstriction. Right-sided valvular heart disease is also a manifestation of the disease.
• Treatment begins with resection of primary tumor with nodal metastases.
• Debulking/metastasectomy is beneficial for controlling symptoms in patients with liver disease or bulky disease.
• Radiation therapy is rarely used for primary therapy, but can be palliative.
• Antihormonal therapy consists of octapeptide analogs of somatostatin; Sandostatin LAR is a helpful, long-acting formulation octapeptide analogs of somatostatin.
• The liver is a common site for metastatic carcinoid, and there are several options for hepatic-directed therapy, including surgery and embolization (chemoembolization or radioembolization).
• Metastatic disease can also be treated with targeted agents or emerging radionuclide therapy.
Pancreatic Neuroendocrine Tumors
• Pancreatic neuroendocrine tumors (NETs) can be sporadic or inherited (MEN).
• Pancreatic NETs are diagnosed on CT/MRI imaging; ultrasound or endoscopic ultrasound (EUS) can help guide biopsy.
• The ENETS staging system is proposed to help stage pancreatic NETs.
• Insulinomas are diagnosed by fasting hypoglycemia with elevated plasma insulin levels; 10% are malignant, and surgical resection (enucleation) is curative.
• Glucagonoma is characterized by migratory necrotizing erythema, insulin-resistant diabetes, glossitis, ileus, and constipation; 50% to 80% are metastatic.
• Because of the higher rate of metastatic disease, surgical resection is curative in less than one-third of patients with glucagonoma.
• Somatostatinoma is characterized by diabetes, diarrhea, and gallbladder disorders.
• Treatment for somatostatinoma includes cytoreductive surgery and chemotherapy.
• Gastrinoma is characterized by ulcer disease in spite of adequate treatment and diarrhea.
• Gastrinoma is diagnosed by hypergastrinemia with elevated basal acid output or positive secretin test; tumors are localized with CT, MRI, or octreotide scan; these tumors are frequently metastatic.
• Targeted therapies such as everolimus and sunitinib have been FDA approved for first-line treatment of pancreatic NETs.
• The incidence of parathyroid carcinoma is 5.73 per 10 million people.
• The etiology of parathyroid cancer has recently been attributed to pericentromeric inversion resulting in overexpression of the cyclin D1 gene.
 In hyperparathyroidism-jaw tumor syndrome, there are mutations in the HRPT2 tumor suppressor gene (parafibromin).
In hyperparathyroidism-jaw tumor syndrome, there are mutations in the HRPT2 tumor suppressor gene (parafibromin).
• Clinical characteristics of parathyroid carcinoma include the constitutional symptoms of primary hyperparathyroidism, including muscle weakness, fatigue, nausea, vomiting, increased thirst, and frequent urination, in addition to bone pain and fractures.
 A neck mass occurs in 34% to 52%, but is uncommon in benign parathyroid adenomas.
A neck mass occurs in 34% to 52%, but is uncommon in benign parathyroid adenomas.
• In parathyroid carcinoma, serum calcium is quite elevated (14.6 to 15.9) with elevated serum parathyroid hormone (PTH) (commonly 10-fold higher than the upper limit of normal).
 The diagnosis is made by laboratory measurement of Ca and PTH, and then 99m-technetium sestamibi scan and neck ultrasound can localize the tumor ± washout for PTH measurement in FNA material.
The diagnosis is made by laboratory measurement of Ca and PTH, and then 99m-technetium sestamibi scan and neck ultrasound can localize the tumor ± washout for PTH measurement in FNA material.
• Pathological features of parathyroid carcinoma include local invasion and lymph node metastases.
• Treatment of parathyroid carcinoma should include en bloc resection of the parathyroid mass with ipsilateral thyroid lobe ± ipsilateral neck dissection followed by postoperative calcium and activated vitamin D supplementation.
• Medical therapy for hypercalcemia precipitated by parathyroid carcinoma should start with hydration and loop diuretics; calcimimetics (Cinacalcet) or bisphosphonates can later be added to lower the serum calcium levels.
• Adjuvant therapy includes chemotherapy, such as dacarbazine, 5-FU, cyclophosphamide; radiotherapy is of limited efficacy.
• Patients with features of hyperparathyroid-jaw-tumor syndrome or a family history should undergo genetic counseling and HRPT2 testing.
• After surgery, one-third of patients are cured, one-third have recurrence after prolonged disease-free survival, and one-third experience a short, aggressive course; the 5-year survival is 83.9%.
Thyroid Cancer
Incidence
Thyroid cancer is the most common endocrine cancer. A spectrum of biological behavior exists, ranging from indolent, well-differentiated tumors to extremely aggressive, poorly differentiated or anaplastic cancers.1,2 Thyroid cancer is the most rapidly increasing malignancy in the United States for both men and women. From 1980 to 2006, the annual U.S. thyroid cancer age-adjusted incidence rose from 4.33 to 11.03 cases per 100,000 population. This is a nearly threefold increase in incidence. The age-adjusted incidence of thyroid cancer is now 11.6 per 100,000 men and women per year.3 This increasing incidence is attributed to improved detection of smaller tumors. Despite this improved detection, the mortality rate remains unchanged at 0.5 per 100,000 population.4,5 Therefore, thyroid cancer presents a unique challenge to the treating physician to manage patient expectations, minimize potentially lifelong complications, employ the appropriate surveillance for followup, and identify patients with more aggressive, poorly differentiated forms.
Classification
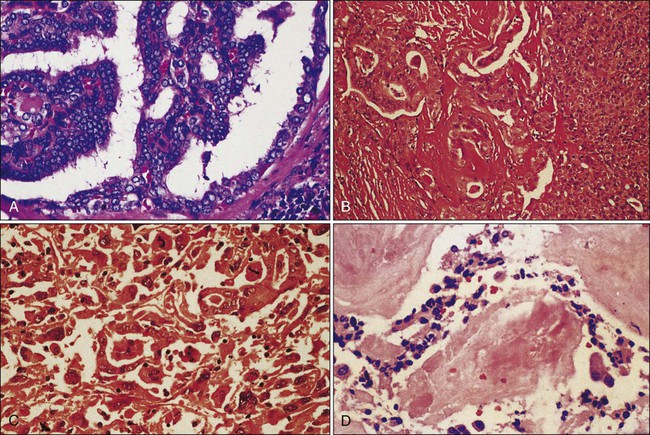
The most common type of thyroid cancer is papillary thyroid cancer (PTC), representing 80% of all cases. The second most common type is follicular thyroid cancer (FTC), which represents 10% to 20% of all cases. Together, papillary and follicular cancers are termed differentiated thyroid cancer (DTC), and both arise from the thyroid follicular cells. Medullary thyroid cancer (MTC) comes from the parafollicular C cells. This neuroendocrine thyroid tumor represents 5% to 10% of all thyroid cancer cases and occurs in familial and sporadic forms. Finally, anaplastic thyroid cancer is one of the most aggressive and rapidly fatal cancers. It can develop from DTC that dedifferentiates over time or it also arises de novo.1,2,6,7 The first part of this section on thyroid cancer considers DTC, and the second part reviews MTC (Table 71-1, Fig. 71-1).
Table 71-1
Histologic Classification of Thyroid Cancers and Their Incidence
| Tumor Histology | Incidence (%) |
| Differentiated carcinomas | 81–87 |
| Papillary | |
| Follicular variant of papillary | |
| Follicular and Hürthle cell | |
| Medullary | 6–8 |
| Anaplastic | 5 |
| Lymphoma | 1–5 |
| Metastatic | <1 |
Etiology
External radiation exposure to the cervical region is one of the most well-known causes of thyroid cancer. Historically, patients received radiation treatments for enlarged tonsils or facial acne. Today, patients with cancer such as Hodgkin disease might still receive radiation treatments. In addition, children exposed to radioactive fallout from the Chernobyl (Russia) accident have demonstrated an increased incidence of thyroid cancer.8,9 Based on evidence from patients radiated for Hodgkin disease, doses of 40 Gy are potentially carcinogenic.10 Epidemiological studies report that 7% to 9% of patients who received 5 to 10 Gy external beam radiation develop thyroid cancer.11 A lag time of 10 to 20 years usually exists between exposure and diagnosis of thyroid cancer, although much shorter periods have been reported (Table 71-2).11
Table 71-2
Risk Factors for Malignancy in Nodular Thyroid
| Low Risk ↔ High Risk | |||||
| Factor | 1 | 2 | 3 | 4 | 5 |
| Age | |||||
| Elderly | • | ||||
| Child | • | ||||
| Sex | |||||
| Male | • | ||||
| Female | • | ||||
| Low-dose radiation in childhood | • | ||||
| Family history | • | ||||
| Cystic mass | • | ||||
| Solid mass | • | ||||
| Multiple masses | • | ||||
| Solitary mass | • | ||||
| Growing mass | • | ||||
| Stable mass | • | ||||
| Hot scan | • | ||||
| Cold scan | • | ||||
| Warm scan | • | ||||
| Fine-needle aspiration (−) | • | ||||
| Fine-needle aspiration (+) | • | ||||
| Associated cervical adenopathy | • | ||||
| Complete resolution in response to thyroid suppression | • | ||||
| Partial resolution in response to thyroid suppression | • | ||||
| No response to suppression | • | ||||
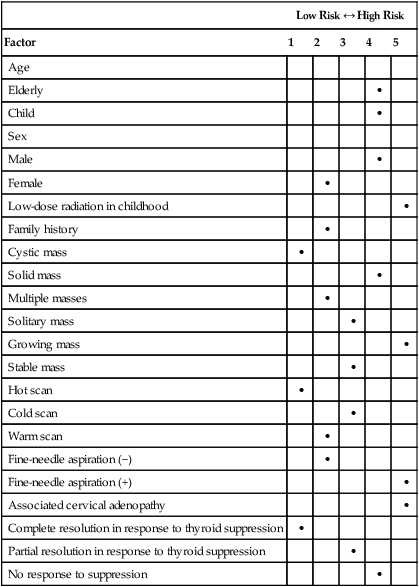
Modified from Sessions RB, Diehi WL. Thyroid cancer and related nodularity. In: Myers E, Suen J, editors. Cancer of the head and neck. 2nd ed. New York: Churchill Livingstone; 1981. p. 766.
Another environmental etiology for thyroid cancer is dietary iodine content. A higher incidence of PTC exists in regions with high dietary iodine content, such as the Pacific rim and Iceland.12 Iodine-deficient countries, in contrast, experience a higher incidence of FTC in addition to benign thyroid goiters. Many factors confound these studies that link changes in DTC rates to iodine intake. Ethnicity, selenium, goitrogen, and carcinogen intake likely play causative roles.13
Increasing investigation into molecular markers that can distinguish carcinoma from benign nodules has led to a greater understanding of the genetic alterations in thyroid cancer. For example, 70% of cancers found in Chernobyl survivors carried a RET and PTC gene (RET/PTC) rearrangement. The fusion of the tyrosine kinase encoding domain of the RET protein with a heterologous group of genes occurs in 20% to 40% cases of PTC and is called the RET/PTC rearrangement.14 RET/PTC rearrangements are frequent in small, multifocal PTCs accompanied by an inflammatory infiltrate, often seen in individuals exposed to ionizing radiation and in children.15 BRAF is a member of the RAF-MEK-ERK serine/threonine kinase-signaling cascade, and a BRAF mutation is found in 40% of PTC cases.16 The V600E BRAF point mutation or mutations in another member of this signaling pathway, RAS, are frequent in cases of poorly differentiated PTC or anaplastic thyroid cancer (ATC).17 Most BRAF substitutions keep the protein in a catalytically active form, resulting in constitutive activation of the RAF-MEK-ERK signaling cascade and constant mitogenic activity.
The Ras proteins are plasma membrane guanosine triphosphatases activated by growth factor receptors. Mutations that result in their constitutive activation lead to oncogenesis. RAS mutations also occur in 20% to 50% of follicular cancers. Similar to the RET/PTC rearrangement, another interchromosomal translocation occurs in FTC. The promoter element of the gene encoding paired box 8 (PAX8) fuses with the coding sequence of the peroxisome proliferator-activated receptor γ (PPARγ) gene in 35% of FTCs.19–19 The functional consequences of the PAX8-PPARγ rearrangement remain unclear. RAS mutations are also highly prevalent in FTC, but Nikiforova et al. found that RAS and PAX8-PPARγ rearrangements are mutually exclusive, suggesting that these are two distinct molecular pathways for FTC development.17
Aside from these acquired genetic lesions, some forms of DTC are also inherited. DTC is seen in familial syndromes such as Gardner syndrome, Cowden syndrome, and Werner syndrome.20,21 Familial nonmedullary thyroid cancer (FNMTC), defined as when two or more first-degree relatives are diagnosed with DTC in the absence of another syndrome, exhibits autosomal-dominant behavior with incomplete penetrance and variable expressivity.23–23 Linkage analyses have identified several candidate genes for FNMTC, including TCO1, MNG1, fPTC/PRN, and NMTC1, but a single responsible gene has not been identified.24–27 Compared to sporadic DTC, FNMTC is more aggressive with increased recurrence, local invasion, multicentricity, and lymph node metastases. However, in the absence of a suitable genetic test, families cannot be screened, and FNMTC is difficult to distinguish from sporadic DTC.28
Classification and Prognosis
DTCs are divided broadly as papillary or follicular. Follicular variant PTC has features of both PTC and FTC, but is classified as a PTC subtype (see Table 71-1 and Fig. 71-1A). In general, well-differentiated PTC has an excellent prognosis with 5-year survival greater than 97%.4 Smaller tumors carry a better prognosis than larger tumors. PTC less than 1 cm in size are called papillary microcarcinomas, and have been reported in 10% to 30% of autopsy studies.31–31 In the past, these tumors were incidentally detected in thyroidectomy specimens, but they are now detected with increasing frequency by high-resolution ultrasound. They are believed to have an excellent prognosis, but some may behave more aggressively than previously appreciated, and management remains controversial.32,33
Age is another important determinant of prognosis in DTC. Older patients tend to have more poorly differentiated, aggressive variants. In these cases, death results from local invasion and extensive metastases. Therefore, the completeness of resection and extrathyroidal extension are two prognostic indicators employed in many staging systems for DTC.34,35
The role of lymph node metastases in determining DTC-specific survival remains controversial. Lymph node involvement is common in PTC, but the exact incidence of lymph node metastases depends on how it is defined. Palpable disease in the lymph nodes is present in 5% to 10% of patients with PTC, but ultrasound detects pathological lymph nodes in 30% of patients. Only 2% of patients with FTC have lymph node metastases since the route of spread is mostly hematogenous, but treatment guidelines and retrospective studies frequently consider PTC and FTC together. Routine histologic examination of lymph nodes detects DTC in 20% to 50% of patients, but when more detailed inspection is performed, up to 90% of patients with DTC will have lymph nodes with microscopic disease. Historically, lymph node involvement was felt to increase local recurrence without affecting survival, and therefore surgeons took a conservative approach to lymph node dissection for DTC. Wada et al. demonstrated that patients with pathologically positive lymph nodes had a recurrence rate of 16.3% compared to 0% in patients without pathological lymph nodes.36 Whether metastatic lymph nodes are evident preoperatively appears to be an important factor determining recurrence. For example, Ito et al. found that if metastatic lymph nodes were not seen preoperatively, then the risk of nodal recurrence was only 1.5%. Of note, in this study of 590 patients with microcarcinomas, 40% of patients had lateral neck lymph node metastases identified histologically after prophylactic neck dissection.37 Hence, lymph node metastases do affect recurrence, and clinically apparent nodes are more important than pathologically positive nodes. The impact of lymph nodes on survival is less clear. Large series and population-based studies suggest that there is a small, but significant, effect on survival.38,39
Because of their questionable effect on mortality, lymph node status is not included in all of the staging systems available for DTC. For example, the AGES system considers age, grade, extrathyroidal extension, and size.40 The AMES system uses age, distant (non-lymph node) metastases, extent of primary tumor, and size.41 Some, like the MACIS (metastases, age, complete excision, invasion, and size) system also account for the adequacy of surgical treatment.42 Alternatively, staging systems developed by the Ohio State University,43 the European Organization for Research and Treatment of Cancer (EORTC),44 the National Thyroid Cancer Treatment Cooperative Study (NTCTCS),45 and the American Joint Committee on Cancer (AJCC)46 all do consider lymph node status. The AJCC staging system is the most widely used (Table 71-3). It is also known as the TNM system because it considers tumor size (T), lymph node metastases (N), and distant metastases (M). Like many of the other thyroid cancer staging systems, it also considers age, with two different classifications for those younger and older than 45 years of age. In those younger than 45 years of age, patients with lymph node metastases are classified as stage I unless they have distant metastases (stage II) (Table 71-4).46
Table 71-3
TNM Classification of Malignant Tumors of the Thyroid Gland
| PRIMARY TUMOR (T STAGE) | |
| Tx | Tumor cannot be assessed |
| T0 | No clinical evidence of tumor |
| T1 | Tumor ≤2 cm limited to the thyroid |
| T2 | Tumor >2 cm and <4 cm limited to the thyroid |
| T3 | Tumor ≥4 cm limited to the thyroid or any tumor with minimal extrathyroid extension |
| T4a | Tumor of any size extending beyond the thyroid capsule to invade the subcutaneous soft tissues, larynx, trachea, esophagus, or recurrent laryngeal nerve |
| T4b | Tumor that invades prevertebral fascia or encases carotid artery or mediastinal vessels |
| ANAPLASTIC CARCINOMAS* | |
| T4a | Intrathyroidal—surgically resectable |
| T4b | Extrathyroidal—surgically unresectable |
| REGIONAL LYMPH NODES (N STAGE) | |
| Nx | Regional nodes cannot be assessed |
| N0 | No palpable nodes |
| N1 | Regional nodal metastases |
| N1a | Level VI nodes (pretracheal, paratracheal, prelaryngeal) |
| N1b | Metastasis to unilateral, bilateral, or contralateral cervical or superior mediastinal nodes |
| DISTANT METASTASES (M STAGE) | |
| Mx | Metastases cannot be assessed |
| M0 | No evidence of distant metastases |
| M1 | Distant metastases present |

*All anaplastic carcinomas are considered T4 tumors.
Adapted from Greene FL, Page DL, Fleming ID, et al, editors. AJCC Cancer Staging Manual. 6th ed. New York: Springer-Verlag; 2002.
Table 71-4
| Stage | TNM |
| PATIENTS < AGE 45 YEARS | |
| I | Any T, any N, M0 |
| II | Any T, any N, M1 |
| PATIENTS ≥ AGE 45 YEARS | |
| I | T1, N0, M0 |
| II | T2, N0, M0 |
| III | T3, N0, M0 |
| T1–3, N1a, M0 | |
| IVA | T4a, N0–1a, M0 |
| T1–4a, N1b, M0 | |
| IVB | T4b, any N, M0 |
| IVC | Any T, any N, M1 |
| MEDULLARY THYROID CANCER | |
| I | |





