Autoimmune and Chronic Cholestatic Disorders of the Liver
Kenneth P. Batts
Introduction
The three most common liver syndromes with a putative autoimmune cause are autoimmune hepatitis (AIH), primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC). Overlap syndromes involve various combinations of these disorders. Other chronic cholestatic disorders discussed in this chapter include hepatobiliary involvement by immunoglobulin G4 (IgG4) disease. Some uncommon noncongenital chronic biliary diseases that can mimic PBC and PSC are reviewed. Pediatric cholestatic disorders (e.g., biliary atresia) are discussed in Chapter 54, and transplantation-related aspects of autoimmune liver disorders are covered in Chapter 52.
Autoimmune Hepatitis
Historically, AIH was called autoimmune-type chronic active hepatitis, but it later evolved to be called autoimmune hepatitis because it is an a priori chronic disease.1 AIH is an autoimmune-mediated syndrome characterized by severe chronic hepatitis that can progress to cirrhosis. It predominantly affects females. It is associated with polyclonal hypergammaglobulinemia, a variety of circulating autoantibodies, an immunogenetic predisposition (i.e., human leukocyte antigen [HLA]-B8, HLA-DR3, or HLA-DR4), absence of viral infection, and a favorable response to immunosuppressive therapy. AIH has been traditionally divided into type 1 (adult) and type 2 (pediatric) (Table 47.1).2
Table 47.1
Subtypes of Autoimmune Hepatitis
| Characteristic | Type 1 | Type 2 |
| Main autoantibody specificity | ANA, ASMA, antiactin | Anti-LKM1, anti-P450 IID6 |
| Genetic predisposition (HLA) | A1, B8, DR3, DR4 | B14, DR3, C4AQ0 |
| Age at onset (usual) | Adult | Pediatric |
| Special features | Lower serum IgG levels Onset more often fulminant May progress rapidly |
|
| Treatment | Corticosteroids | Corticosteroids |
ANA, Antinuclear antibody; anti-LKM1, anti–liver-kidney microsomal antibody 1; anti-P450 IID6, anti–cytochrome 450 IID6; ASMA, anti–smooth muscle antibody; HLA, human leukocyte antigen; IgG, immunoglobulin G.
The major symptoms and findings of AIH were initially incorporated into a scoring system by the International Autoimmune Hepatitis Group in 1992; the total score describes the probability of a patient having AIH.1 Subsequent updated3 and simplified4 versions exist. In these systems, a combination of clinical and laboratory features is used to predict the likelihood that a patient with chronic hepatitis has AIH. The original system had a high sensitivity for AIH but a low specificity for distinguishing AIH from PBC, which prompted the development of a revised system, which is summarized in Table 47.2.3,4
Table 47.2
Diagnostic Criteria for Autoimmune Hepatitis: Minimum Required Parameters
| Parameters | Score |
| History | |
| Sex | |
| Female | +2 |
| Male | 0 |
| Alcohol average consumption | |
| <25 g/day | +2 |
| >60 g/day | −2 |
| History of hepatotoxic drug use | |
| Yes | −4 |
| No | +1 |
| Other autoimmune diseases | |
| Present | +2 |
| Absent | 0 |
| Serum Biochemical Studies | |
| Serum ALP/ALT ratio | |
| <1.5 | +2 |
| >3.0 | −2 |
| Serum total globulin, γ-globulin, or IgG | |
| >2 × normal | +3 |
| 1.5-2 × normal | +2 |
| 1.0-1.5 × normal | +1 |
| Normal | 0 |
| Serology Studies | |
| ANA, ASMA, or LKM1 | |
| >1 : 80 | +3 |
| 1 : 80 | +2 |
| 1 : 40 | +1 |
| <1 : 40 | 0 |
| AMA | |
| Negative | 0 |
| Positive | −4 |
| Viral hepatitis markers | |
| Negative | +3 |
| Positive | −4 |
| Other autoantibodies | |
| Present | +2 |
| Absent | 0 |
| HLA-DR3 or -DR4 | |
| Present | +1 |
| Absent | 0 |
| Treatment Response | |
| Complete remission | +2 |
| Remission with subsequent relapse | +3 |
| Histology | |
| Interface hepatitis | +3 |
| Lymphoplasmacytic infiltrate | +1 |
| Hepatocyte rosetting | +1 |
| None of the above | −5 |
| Biliary changes | −3 |
| Other changes | −3 |
| Interpretation of Aggregate Scores | |
| Before therapy | |
| >15 | Definite AIH |
| 10-15 | Probable AIH |
| After therapy | |
| >17 | Definite AIH |
| 12-17 | Probable AIH |
AIH, Autoimmune hepatitis; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AMA, antimitochondrial antibody; ANA, antinuclear antibodies; ASMA, anti–smooth muscle antibody; HLA, human leukocyte antigen; LKM1, liver-kidney microsomal antibody 1.
From Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Knowledge of the pertinent clinical and laboratory parameters during interpretation of a liver biopsy sample is extremely helpful. Factors that favor AIH are female sex, a hepatitic rather than a cholestatic liver enzyme profile (i.e., serum aminotransferases are elevated more prominently than alkaline phosphatase), hypergammaglobulinemia, serum autoantibodies (see Pathogenesis), and absence of evidence of virus-, drug-, or alcohol-related liver disease. A clinical and biochemical response to immunosuppressive therapy is also an important criterion.3 However, independent of a biopsy, this scoring system should be interpreted with caution. Although useful in excluding AIH (because few patients with diseases other than AIH have scores that fall into the probable AIH category5), exceptions may occur for patients with nonalcoholic fatty liver disease.6
Clinical Features and Epidemiology
The incidence of classic (type 1) AIH is 1.9 cases per 100,000 individuals per year, which is a higher rate than for PBC or PSC. Patients of any age may be affected. Concurrent autoimmune diseases of other types are common (34% of women and 17% of men), and 78% of AIH patients are women.7 A subtype of AIH, characterized by the presence of anti–liver-kidney microsomal antibody (anti-LKM), is sometimes referred to as AIH type 2.8 It is more common among children and females than males.8
Individuals with certain human leukocyte antigen (HLA) types are at increased risk for AIH, although testing for the various phenotypes is not necessary in routine clinical practice. The HLA-A1, HLA-B8, HLA-DR3, and HLA-DR4 phenotypes are associated with type 1 AIH.1,8
In patients with AIH, serum biochemical values vary considerably, which is a reflection of the grade and stage of disease. During periods of inactive (quiescent) disease, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values may be normal. However, during disease flare-ups, the serum aminotransferase values may range from the low hundreds to an excess of 1000 IU/mL. Serum alkaline phosphatase values usually are normal or only minimally elevated. Bilirubin values are often normal but may be elevated during bouts of clinical activity. Hypergammaglobulinemia, which is usually evident even in early-stage disease, is a defining feature of AIH.1 Cases of AIH that progress to cirrhosis have elevations of bilirubin levels and biochemical evidence of cirrhosis (e.g., thrombocytopenia, decreased serum albumin, prolonged prothrombin time).
Pathogenesis
Similar to other types of autoimmune diseases, AIH is caused by a defect in suppressor T cells that leads to disordered immunoregulation and production of a variety of autoantibodies, some of which probably act against hepatocyte surface antigens. The defect in suppressor T cells may occur de novo (most common) or after a triggering episode. Hepatitis A10 and a variety of drugs11 are among the best described inciting factors. Although most of the autoantibodies in AIH patients are not specific for AIH and may not act directly in the pathogenesis of the disease, their identification is often helpful diagnostically.
Antinuclear antibodies (ANAs) are identified in approximately 80% of patients with AIH and typically occur in a titer greater than 1 : 40. Anti–smooth muscle antibody (ASMA) is positive in about 70% of AIH patients, also occurring in a titer greater than 1 : 40. The finding of high-titer ANA or ASMA, or both, in a patient with clinical suspicion of AIH defines the type 1 form of this disorder, which is the most common and represents the prototypical type. However, ANA and ASMA may be identified in the serum of patients without AIH and in patients with other types of liver diseases, such as hepatitis C, alcoholic liver disease, or nonalcoholic steatohepatitis, but in these cases, the titer is usually low.
The presence of anti-LKM is a characteristic feature of type 2 AIH, and it is usually detected in the absence of high-titer ANA or ASMA. High-titer anti-LKM in the absence of anti–hepatitis C virus antibody (anti-HCV) is designated type 2 AIH.12 This uncommon form of AIH occurs most often in children. Low-titer anti-LKM with anti-HCV is designated type 2b AIH by some, although it probably does not reflect an important pathologic entity. This type of AIH is more appropriately viewed as a variant of hepatitis C, which also has autoimmune features. Antibodies against soluble liver antigen (SLA), liver-pancreas (LP) antigen, and asialoglycoprotein receptor (ASGPR) may also be identified in type 1 AIH, but they are less diagnostically useful.13,14
Although the precise mechanism of immune attack on the liver remains unknown, based on the histopathology, it is postulated that hepatocytes, rather than biliary epithelial cells, are the primary target. Necrosis of hepatocytes is usually most prominent at the level of the limiting plate (i.e., zone 1, periportal hepatocytes). This process is also referred to as piecemeal necrosis, interface hepatitis,15 or troxis hepatitis.16 A predominant attack on hepatocytes in zone 3 (pericentral) occurs in some cases.17 Necrosis of random hepatocytes throughout the hepatic lobules is also common in AIH and is recognizable as acidophil bodies or their remnant, lipofuscin-laden macrophages.
Pathology
Gross examination of the liver in AIH contributes little diagnostically. In early-stage disease, the liver usually appears normal. If submassive or massive hepatocyte necrosis occurs during episodes of severe activity, the external surface of the liver may appear shriveled, and cross sections may show areas of confluent parenchymal collapse. While the disease progresses, cirrhosis eventually develops, and it is typically composed of tan to brown micronodules and macronodules (Fig. 47.1), similar to the pattern that occurs in chronic viral hepatitis. Occasionally, a greenish discoloration may be seen, similar to cirrhosis resulting from chronic biliary disorders.

Although the pathologic lesions in AIH vary considerably according to the grade and stage of disease (Table 47.3), chronic inflammation composed primarily of lymphocytes and a considerable number of plasma cells in portal tracts is a near-constant microscopic feature. However, during periods of quiescent disease (i.e., remission), portal inflammation may be the only histologic abnormality. In some cases, little or no inflammation is detected (Fig. 47.2). A small number of intraepithelial lymphocytes may be found within bile duct epithelium. However, bile duct destruction and loss are not features of AIH and should alert the pathologist to a possible diagnosis of PBC or an overlap syndrome.
Table 47.3
Comparative Histopathology of Necroinflammatory Features in Autoimmune Hepatitis, Primary Biliary Cirrhosis, and Primary Sclerosing Cholangitis
| Staging | Inflammation | Necrosis | |||
| Portal | Periportal | Lobular | Interface | Lobular | |
| Autoimmune Hepatitis | |||||
| Grade 0 | 0-2+ | 0 | 0-1+ | 0 | 0 |
| Grade 1 | 1-3+ | 1+ | 1-2+ | ≤1+ | ≤1+ |
| Grade 2 | 2-4+ | 1-2+ | 1-2+ | ≤2+ | ≤2+ |
| Grade 3 | 2-4+ | 2-3+ | 2-3+ | ≤3+ | ≤3+ |
| Grade 4 | 2-4+ | 2-4+ | 3-4+ | ≤4+ | ≤4+ |
| Primary Biliary Cirrhosis | |||||
| Stage I | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage II | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage III | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Stage IV | 2-4+ | ≤1+ | 1+ | 0 | 0 |
| Primary Sclerosing Cholangitis | |||||
| Stage I | 1-2+ | ≤1+ | 0 | 0 | 0 |
| Stage II | 1-3+ | ≤1+ | 0 | 0 | 0 |
| Stage III | 1-3+ | ≤1+ | 0 | 0 | 0 |
| Stage IV | 1-3+ | ≤1+ | 0 | 0 | 0 |
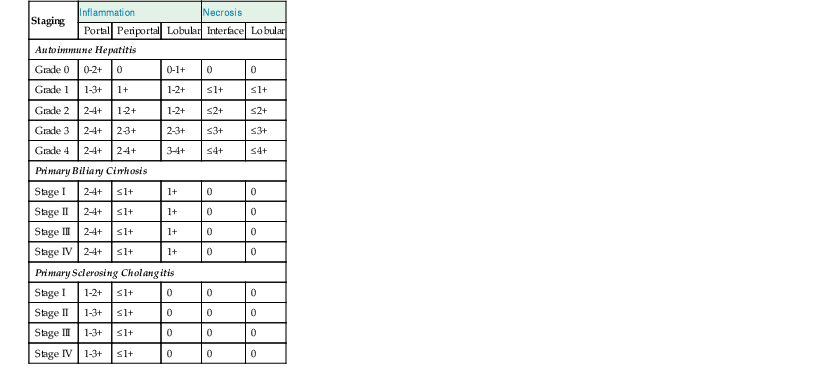
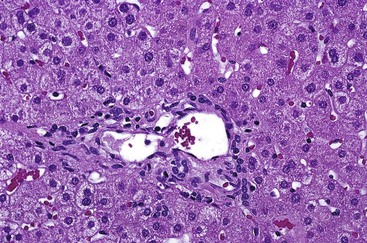
During flares of disease activity, interface hepatitis, which is the pathologic hallmark lesion of active AIH, is typically prominent. This lesion is characteristic of AIH, but it is not specific. Interface hepatitis is also common in viral- and drug-associated forms of chronic hepatitis, in PBC, and in other diseases. Interface hepatitis in AIH is characterized by a prominent lymphohistiocytic infiltrate at the portal tract mesenchymal-parenchymal junction with accompanying histologic evidence of liver cell damage (Fig. 47.3, A). CD8-positive T cells are a dominant subset of lymphocytes within areas of interface hepatitis, and CD4-positive T cells predominate within the portal tracts.18
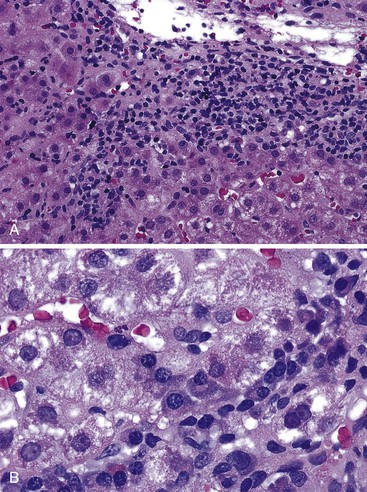
Damage caused by interface hepatitis may appear as overt necrosis of whole hepatocytes (i.e., acidophil bodies) or as slow destruction of hepatocytes caused by ingestion of small portions of cytoplasm by activated lymphocytes, described vividly by Wang and associates as troxis, which is the Greek word for “gnaw” or “chew.”16 In AIH, plasma cells are normally quite conspicuous, particularly compared with other disorders such as chronic viral hepatitis (see Fig. 47.3, B).
In addition to portal and periportal inflammation, lobular damage usually occurs in AIH. It manifests as some degree of anisonucleosis, ballooning degeneration, hepatocellular swelling, neocholangiole formation, cholestasis (rare), and acidophil bodies accompanied by a lymphocytic and plasmacytic infiltrate (Fig. 47.4). The degree of lobular damage contributes to the grade of disease. Although hepatocellular damage is usually panlobular, in some cases, zone 3 (centrilobular) damage may be prominent,16 resembling periportal interface hepatitis but occurring in the centrilobular zones of the liver (Fig. 47.5). This may represent an early form of AIH that precedes overt portal-dominant (classic) AIH.19
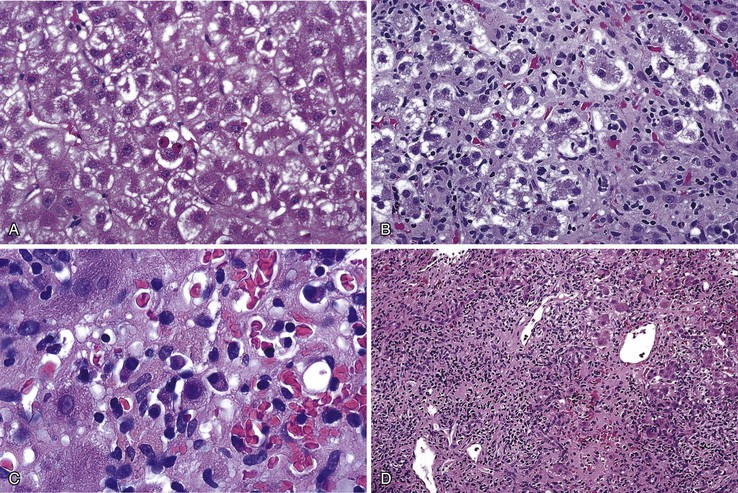
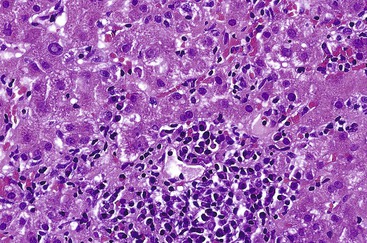
Lobular hepatitis is typically prominent during bouts of disease activity, but it may resolve spontaneously or as a result of immunosuppression. The activity grade can be assessed semiquantitatively, similar to hepatitis C. However, compared with chronic hepatitis C, AIH may have a much broader range of disease activity, from complete inactivity at one end of the spectrum to submassive or massive necrosis with confluent necrosis (see Fig. 47.4) at the other. Liver sampling is only rarely performed immediately after treatment, but rapid resolution of lobular necrosis with complete or incomplete resolution of portal inflammation and resolution of fibrosis is typical.
Cholestasis is usually not a feature of AIH, but a mild degree may be seen in more severe cases with marked lobular inflammation (Fig. 47.6). In contrast to hepatitis C, steatosis is not a typical feature of AIH. Lymphocytic venulitis involving portal or central veins may occur in high-grade disease, but it does not usually result in vein destruction or substantial endothelial damage, as in acute allograft rejection.
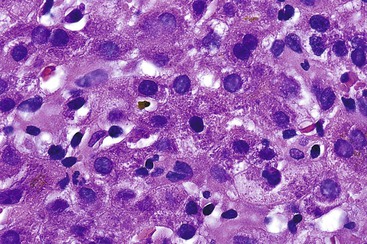
Similar to chronic viral hepatitis, progression of fibrosis (stage) from portal to initially periportal and then to bridging fibrosis and cirrhosis occurs in AIH. Even after cirrhosis has developed, interface hepatitis may persist within fibrous septa or regenerative nodules (i.e., active cirrhosis). Alternatively, the inflammation may subside completely, resulting in inactive cirrhosis.
Although AIH is a chronic disease, onset occurs at some point, and the disease may be regarded as acute. Relatively little is known regarding the pathology of acute-onset AIH. Lefkowitch and colleagues reported hepatocyte swelling, acidophil bodies, steatosis, cholestasis, piecemeal necrosis, and prominent portal plasma cells in initial liver biopsies from two patients with clinical features of AIH.20 Another study that examined liver samples from patients with clinical disease of less than 6 months’ duration found that with the exception of one case of lobular hepatitis with confluent necrosis (i.e., submassive hepatic necrosis), 25 of 26 of patients already had histologic changes of chronicity, such as portal inflammation and fibrosis (7 of 26) or bridging fibrosis (18 of 26).21 This suggests that most cases of clinically acute AIH represent a flare of previously occult chronic disease. This idea was further supported by observations in a related study that found that patients with AIH of less than 3 months’ duration were indistinguishable by clinical and laboratory tests from patients with disease of at least 12 months’ duration.22 These findings support the view that AIH is an a priori chronic disease.1
Differential Diagnosis
Acute-Onset Autoimmune Hepatitis
The histologic differential diagnosis of AIH is rather broad and depends on the clinical context of the affected patient. For patients who have severe, clinically acute hepatitis with prominent hepatocyte necrosis, the major differential diagnostic considerations include severe drug or toxic injury, fulminant A or B hepatitis, Epstein-Barr virus (EBV) (rare), and other types of systemic viral infection (see Table 47.3).
Distinguishing AIH from drug- or toxin-associated hepatitis can usually be accomplished by applying the principles of the scoring system of Alvarez and associates3 (see Table 47.2), which uses autoantibody status and hypergammaglobulinemia to establish a correct diagnosis. A thorough clinical history regarding use of drugs or toxins is essential.
In equivocal cases, a rapid response to immunosuppressive therapy helps support a diagnosis of AIH. Serologic testing for hepatitis A virus (HAV), hepatitis B virus (HBV), and EBV is usually quite reliable for distinguishing AIH and acute viral hepatitis. Acute hepatitis C is rarely a diagnostic concern, but when suspected, a positive serum assay result for hepatitis C virus (HCV) RNA is the best available diagnostic tool because anti-HCV antibodies may occur after the hepatitic episode. An initial absence of anti-HCV with subsequent development within weeks helps to support a diagnosis of acute hepatitis C infection.
Chronic Autoimmune Hepatitis
The pathology associated with typical AIH (i.e., classic pattern of chronic active hepatitis) may resemble chronic HBV infection with or without delta virus superinfection, hepatitis C, PBC, PSC, or chronic drug-related hepatitis (Table 47.4).
Table 47.4
Differential Diagnosis of Autoimmune Hepatitis
| Disease Pattern | Possible Diagnosis |
| Acute-onset pattern of predominant lobular hepatitis |
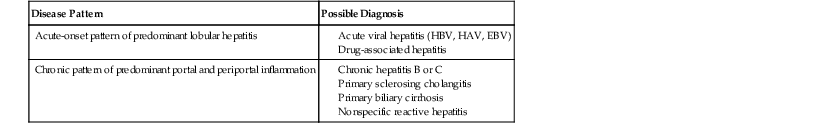
EBV, Epstein-Barr virus; HAV, hepatitis A virus; HBV, hepatitis B virus.
A diagnosis of chronic viral hepatitis is most often established serologically. However, the morphologic features may be helpful (Table 47.5). Brisk (grade 3 or 4) active chronic hepatitis usually excludes HCV, which almost always exhibits only minimal to mild disease, with transaminase levels that usually do not exceed 200 to 300 IU/mL. Occasionally, chronic HCV may exhibit fairly prominent plasma cell infiltrates. This has been designated by Czaja and associates as a form of chronic hepatitis C with autoimmune features.23 It likely accounts for examples of AIH type 2a (described earlier).
Table 47.5
Differentiation of Autoimmune Hepatitis from Chronic Viral Hepatitis
| Diseases | Helpful Hints |
| AIH vs. HCV |

AIH, Autoimmune hepatitis; HbcAg, hepatitis B core antigen HbeAg, hepatitis B e antigen; HbsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus.
Data from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Chronic HBV infection may exhibit considerable disease activity that can mimic flares of AIH, but it is usually in the context of superimposed delta virus infection or emergence of a mutant strain of HBV. Prominent plasma cells help to distinguish AIH from chronic HBV infection, but the serologic profile is the most reliable means of ruling out chronic HBV infection.
Differentiating AIH from PBC and PSC is usually straightforward histologically, serologically, and biochemically (i.e., cholestatic enzyme pattern in PBC and PSC versus a hepatitic pattern in AIH) (Table 47.6; see also Table 47.3). The overlap syndromes of AIH and PBC and of AIH and PSC are discussed later in this chapter. Although PBC and PSC may demonstrate spillover of lymphocytes across the limiting plate into zone 1, neither disease demonstrates overt hepatocyte necrosis to any significant degree. In some cases, the number of portal plasma cells in PBC may be similar to that in AIH. However, PSC usually has fewer plasma cells. Although AIH, PBC, and PSC may have lymphocytes within bile duct epithelium (i.e., lymphocytic cholangitis), destruction of ducts and ductopenia are rare in AIH.24 Florid duct lesions characteristic of PBC and fibrous obliterative lesions characteristic of PSC are not seen in AIH, except in overlap syndromes.
Table 47.6
Differentiation of Autoimmune Liver Diseases
| Diseases | Helpful Hints |
| AIH vs. PBC |

AIH, Autoimmune hepatitis; PBC, primary biliary cirrhosis; PSC, primary sclerosing cholangitis.
Data from Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-938.
Grading and Staging
In liver pathology, the diagnosis should be rendered based on the cause whenever possible. For AIH, the grade of inflammatory activity should be reported. The simplest system for grading activity in AIH separates it into none, minimal, mild, moderate, and severe, which corresponds to a numerical grade of 0 through 4, respectively.12,25,26 This system is based primarily on the degree of piecemeal necrosis and lobular necrosis (see Table 47.3). If there is a discrepancy between these two patterns of necrosis, the more severe should prevail. The French METAVIR system devised for chronic viral hepatitis is similar, but it uses a scale of 0 to 3.27 The Knodell scoring system28 is useful for scientific studies, but its main drawback is that it does not distinguish necroinflammatory activity (i.e., grade) from fibrosis (i.e., stage). This problem was rectified in an updated version of this system published by Ishak and coworkers in 1995.29
An assessment of the degree of fibrosis (i.e., stage) should be reported. A variety of schemes exist, and the choice of a system is best left to the discretion of the pathologist based on input from the clinicians. The simplest system is based on a 0 to 4 scale that corresponds to absent, portal, periportal, bridging fibrosis, and cirrhosis, respectively (Table 47.7).12,25,26 The French METAVIR system is similar, but it uses a 0 to 3 scale.27 The updated Knodell system uses a 0 to 6 scheme.29

Natural History and Treatment
Because most cases of AIH respond to immunosuppressive therapy, the response to therapy can be included in the AIH scoring system.3 Initial treatment mainly uses corticosteroids with or without azathioprine. Eventually, many patients may be weaned from corticosteroids and maintained on azathioprine alone; 10% to 20% can eventually be weaned from all types of immunosuppression. Clinicians are often reluctant to discontinue immunosuppression if the serum transaminase levels remain elevated by more than two times normal or if histologic disease activity (grade 1 or greater in a four-grade system) persists.30 In cases that are steroid refractory or in which steroid side effects are an issue or azathioprine intolerance exists, cyclosporine, tacrolimus, or mycophenolate mofetil can be effective alternatives.31 For cases that progress to cirrhosis, liver transplantation is an acceptable form of therapy, although AIH may recur within the allograft.32
Primary Biliary Cirrhosis
PBC is a chronic bile duct–destructive disease that results in progressive cholestasis and cirrhosis if left untreated. Although the precise pathogenesis remains uncertain, considerable evidence indicates that it is an autoimmune disease that occurs in genetically predisposed individuals after stimulation by an environmental factor.33 PBC is associated with a variety of nonhepatic autoimmune diseases and elevated levels of serum autoantibodies.
The term PBC is commonly used even though it describes only the last stage of disease. A more appropriate term is the syndrome of PBC or chronic nonsuppurative destructive cholangitis.34,35 Although the latter term is fitting, it may also apply to other conditions, such as irreversible hepatic allograft rejection. Nevertheless, the term PBC is thoroughly entrenched in the literature and is likely to remain the term of choice by most clinicians. A PBC-like illness in patients who lack elevated levels of serum antimitochondrial antibodies (AMAs) (e.g., autoimmune cholangitis) is discussed later.
Clinical Features and Epidemiology
Approximately 90% of patients with PBC are female and between the ages of 40 and 60 years (range, 20 to 80 years). PBC occurs worldwide, and all races are susceptible. There is some familial clustering of PBC, with daughters of affected mothers at highest risk, but overall, less than 1% of first-degree relatives of patients with PBC develop the disorder.36 In a mostly white population in Olmsted County, Minnesota, the age-adjusted incidence of PBC is 4.5 and 0.7 per 100,000 women and men, respectively, with corresponding prevalence rates of 65.4 and 12.1 per 100,000.37
PBC coexists with other autoimmune diseases, with the strongest associations with connective tissue disorders such as rheumatoid arthritis, CREST syndrome (calcinosis, Raynaud disease, esophageal dysmotility, sclerodactyly, and telangiectasia), systemic lupus erythematosus, dermatomyositis, interstitial lung disease, and autoimmune thyroid disease.38 An association between celiac disease and PBC has been described.39 However, these associations most likely represent clustering of diseases in patients who are prone to autoimmune conditions rather than a direct pathogenetic link.
In early stages, PBC is usually asymptomatic and is detected only by identification of abnormal serum liver test results. In early disease, elevated serum levels of alkaline phosphatase and γ-glutamyltransferase (GGT) are typical. Serum cholesterol levels are usually elevated, and serum bilirubin levels are usually normal or only minimally elevated (<2 mg/dL). Serum ALT and AST levels are usually normal or only mildly elevated. Serum immunoglobulin M (IgM) levels may be mildly elevated.
In more advanced disease, the same general biochemical patterns persist. However, bilirubin levels tend to rise progressively, and the biochemical stigmata of cirrhosis (i.e., decreased serum albumin, prolonged prothrombin time, and thrombocytopenia) eventually become apparent. In later stages, the signs and symptoms of disease may be divided into those related to progressive cholestasis and those related to cirrhosis. Among the former are pruritus, xanthomas, jaundice, and osteoporosis. The signs and symptoms of cirrhosis in PBC are the same as for other causes of cirrhosis: esophagogastric varices, ascites, spider angiomas, and splenomegaly.
Pathogenesis
There is strong evidence that PBC represents an autoimmune attack directed toward autoantigens on biliary epithelium. PBC is thought to be initiated when tolerance to the dihydrolipoyl transacetylase (E2) subunit of the pyruvate dehydrogenase complex (PDC-E2) is lost, resulting in development of PDC-E2 specific–AMAs.33,40 The most conspicuous histologic features of PBC—lymphocytic and granulomatous destruction of intralobular and septal bile ducts—are tied to pathogenesis through the observation that intact immunoreactive PCD-E2 exists within apoptotic blebs of biliary epithelial cells.41 This protein has been demonstrated on allograft biliary epithelium in patients with recurrent PBC after liver transplantation.42
Although characteristic of PBC, AMAs are detected by using standard immunofluorescence techniques in only 92% to 95% of patients with clinical and histologic features of PBC, and they can be seen in patients without the typical clinical and histologic features of this disorder.43 ANAs are seen in approximately one third of patients, and the ANAs tend to be associated with a more rapid progression of disease.44 Approximately 5% to 8% of PBC patients lack AMAs (i.e., AMA-negative PBC), also known as autoimmune cholangitis (AC).
The type of inflammation, the lymphocytic and granulomatous duct destruction, the associated autoimmune diseases of other organs, and occurrence of overlap syndromes of PBC and AIH provide strong support of PBC as an autoimmune disorder. The precise pathogenesis remains unknown. However, T cell–mediated cytotoxicity and molecular mimicry after infection likely play a central role.33 Some have proposed a role for endothelin overproduction and biliary ischemia in the development of PBC.45
Pathology
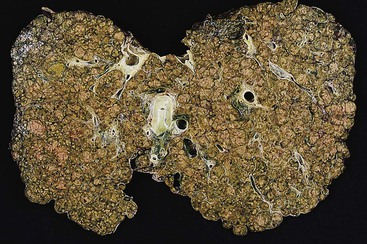
In early-stage PBC, the liver is often slightly enlarged and variably bile stained. In later stages, macronodular cirrhosis develops and is often associated with an intense green hue that reflects progressive cholestasis (Fig. 47.7). In contrast to PSC, cholangiectases and cholangitic abscesses are not seen.
The hallmark lesion of PBC is destructive cholangitis that affects interlobular and septal bile ducts and results in duct loss and subsequent biliary cirrhosis. The term nonsuppurative destructive cholangitis accurately describes these lesions, but the more succinct term florid duct lesion is more commonly used.46 Because these lesions are usually focal, they may be missed on liver biopsy. The florid duct lesion is characterized by a portal lymphocytic infiltrate and epithelioid cells centered on septal and interlobular bile ducts, with histologic evidence of bile duct damage and destruction. Characteristic features include disruption of the bile duct basement membrane, intraepithelial lymphocytes and plasma cells, biliary epithelial cytoplasmic vacuolization, regenerative hyperplasia, and occasional mitotic figures. Granulomas are usually identified.
Florid duct lesions may reflect granulomatous duct destruction (Fig. 47.8) or more severe examples of lymphocytic cholangitis (Fig. 47.9
Related posts:
 Diagnostic Cytology of the Biliary Tract and Pancreas
Diagnostic Cytology of the Biliary Tract and Pancreas
 Algorithmic Approach to Diagnosis of Inflammatory Disorders of the Gastrointestinal Tract
Algorithmic Approach to Diagnosis of Inflammatory Disorders of the Gastrointestinal Tract
 Epithelial Neoplasms of the Stomach
Epithelial Neoplasms of the Stomach
 Polyps of the Stomach
Polyps of the Stomach
 Benign and Malignant Tumors of the Gallbladder and Extrahepatic Biliary Tract
Benign and Malignant Tumors of the Gallbladder and Extrahepatic Biliary Tract
 Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
