CHAPTER 65 Arrhythmogenic Right Ventricular Dysplasia
DEFINITION
ARVD is a genetic cardiomyopathy principally involving the right ventricle that is characterized clinically by ventricular arrhythmias and progressive structural alterations in right ventricular size and function. Fibrofatty replacement of the right ventricular myocardium forms the histopathologic hallmark of this disease. There are three patterns of disease expression: (1) classic, characterized by disease limited to the right ventricle with either no or minimal involvement of the left ventricle, which appears late in the course of the disease; (2) left dominant, characterized by early or predominant left ventricular involvement; and (3) biventricular, characterized by parallel involvement of both ventricles from the beginning of disease process.1
PREVALENCE AND EPIDEMIOLOGY
Although the exact incidence of ARVD is unknown, the prevalence has been estimated to be 0.02% of the general population.2 ARVD reportedly is more prevalent (0.8%) in certain parts of northern Italy (Veneto) and Greece (Naxos Island).3,4 ARVD most commonly manifests after puberty and before age 50 years.5,6 ARVD is more common in highly athletic individuals. It is an important cause of sudden cardiac death in young individuals, particularly in athletes, accounting for 5% of sudden deaths in individuals younger than 35 years in the United States, and 25% of deaths in athletes in the Veneto region of Italy.7 ARVD is believed to be more common than reported because of its frequently asymptomatic clinical course, difficulty in diagnosis using conventional noninvasive methods, and frequent misdiagnosis.
ETIOLOGY AND PATHOPHYSIOLOGY
The exact pathogenesis of ARVD remains speculative, but it is most widely believed to be due to genetic abnormalities mainly affecting cardiac desmosomal structure and function. In the United States, the most common reported genetic abnormality in ARVD is a mutation in plakophillin-2 (PKP2), which is a desmosomal protein and is present in more than one third of patients. Desmosomal mutations are believed to disrupt cell-cell adhesion, which provokes myocyte detachment and death under conditions of high mechanical stress, as suggested by its frequent occurrence in athletics and the thinnest portions of the right ventricle, such as the inferior subtricuspid region, right ventricular apex, and right ventricular infundibulum—together termed the triangle of dysplasia (Fig. 65-1).8 This leads to a compensatory repair process characterized by inflammation and fibrofatty substitution. Familial ARVD accounts for 30% to 80% of cases, and most commonly shows an autosomal dominant inheritance, with age-dependent penetrance and variable clinical expression, which can be attributed to the complex interplay of age, gender effects, and environmental influences such as exposure to viruses and athletic activity with the underlying genetic background in the ultimate disease causation and progression. Mere inheritance of a genetic abnormality does not imply “disease” for the same reason. To date, at least 11 distinct subtypes of autosomal dominant ARVD are known, and at least seven genes have been implicated in ARVD (Table 65-1).9
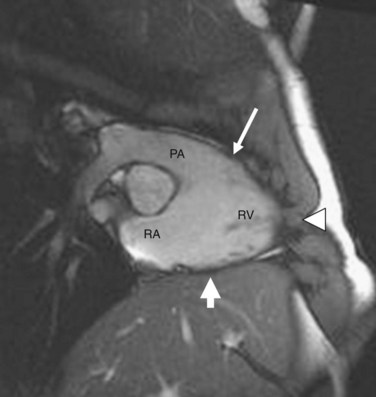
 FIGURE 65-1
FIGURE 65-1
Genetic mutations are also implicated in determining the ventricular predominance in ARVD. Chain-termination mutations causing truncation of the C-terminal domain of desmoplakin can disrupt cytoskeletal integrity, predisposing to predominant left ventricular involvement. Conversely, defects in the N-terminal domain of desmoplakin can result in a primary dysfunction of cell adhesion, leading to predominant right ventricular disease.8
In addition to genetic causes, other important proposed etiologic mechanisms of ARVD are (1) apoptosis (programmed cell death), (2) inherited metabolic or ultrastructural defects leading to myocardial dystrophy, and (3) secondary complication of a prior primary right ventricular myocarditis on the basis of frequently found myocardial inflammatory infiltrates and isolation of coxsackie B and enteroviral RNA viruses in some biopsy samples.10
Pathologically, ARVD is characterized by gradual myocyte loss and fibrofatty infiltration, which starts in the subepicardial region and gradually involves the endocardium, often sparing the trabecular myocardium. Plexiform arrangement of the residual myocardial fibers, separated by fat and fibrosis, leads to electrical instability and provides a conduit for a reentrant phenomenon. This situation explains the arrhythmogenic propensity characteristic of this disease, which can be worsened further by superimposed myocarditis that occurs frequently in ARVD because of the genetic susceptibility.11 A myocardial dysplastic process and the associated loss of its strength and integrity are responsible for the varying degrees of structural derangement and functional disturbances of the heart seen in ARVD.
MANIFESTATIONS OF DISEASE
Clinical Presentation
ARVD usually manifests between the second and fifth decades (mean age at diagnosis approximately 30 years). Patients most commonly present with symptomatic ventricular arrhythmias or sudden cardiac death, or may remain asymptomatic, with ARVD discovered only incidentally on routine examination. The most common symptoms are palpitations, syncope, atypical chest pain, and dyspnea. Ventricular arrhythmias are often precipitated by exercise or stress, and range from isolated ventricular premature beats to sustained monomorphic or polymorphic ventricular tachycardia with left bundle branch block morphology, or abrupt ventricular fibrillation, which is the most common underlying cause of sudden cardiac death in ARVD.12
In addition to ventricular arrhythmias, about 25% of patients may also show supraventricular arrhythmias, which include atrial fibrillation, atrial tachycardia, and atrial flutter, in that order of frequency.13 Left ventricular involvement is associated with an increased incidence of arrhythmic events, more severe right ventricular wall thinning, and heart failure. Frank left-sided heart failure is rare. Figures 65-2, 65-4, and 65-6 illustrate left ventricular involvement in ARVD.
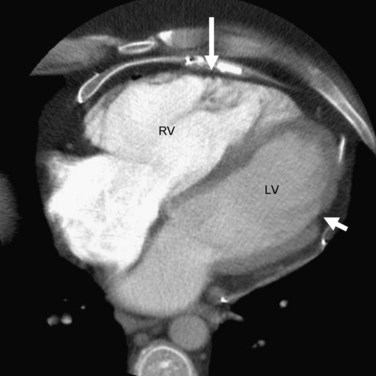
 FIGURE 65-2
FIGURE 65-2Diagnosis
Diagnosis of ARVD is challenging and is currently based on family history and morphofunctional, ECG, histopathologic, and clinical findings as proposed by the Task Force of Cardiomyopathies in 1994 (Table 65-2).14 The diagnosis is made in the presence of two major criteria, one major criterion and two minor criteria, or four minor criteria. Various modifications of the existing Task Force criteria are being increasingly proposed and emphasize the increment in the diagnostic sensitivity of these criteria for less severe forms of the disease and asymptomatic first-degree relatives of affected probands. These modifications include more comprehensive characterization of ECG and echocardiographic criteria, inclusion of mutation analysis, and descriptions of left ventricular involvement.15,16
TABLE 65-2 Task Force Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Dysplasia
| Major Criteria | Minor Criteria | |
|---|---|---|
| Global or regional dysfunction and structural alterations* | Severe dilation and reduction of right ventricular ejection fraction with no (or only mild) left ventricular impairment | Mild global right ventricular dilation or ejection fraction reduction with normal left ventricle |
| Localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging) | Mild segmental dilation of right ventricle | |
| Severe segmental dilation of right ventricle | Regional right ventricular hypokinesia | |
| Tissue characterization of wall | Fibrofatty replacement of myocardium on endomyocardial biopsy specimen | |
| Repolarization abnormalities | Inverted T waves in right precordial leads (V2 and V3) in individuals >12 yr old in absence of right bundle branch block | |
| Depolarization or conduction abnormalities | Epsilon waves or localized prolongation (>110 ms) of the QRS complex in right precordial leads (V1-V3) | Late potentials (signal-averaged ECG) |
| Arrhythmias | Left bundle branch block ventricular tachycardia (sustained and nonsustained) by ECG, Holter, or exercise testing | |
| Frequent ventricular extrasystoles (>1000/24 hr) (Holter) | ||
| Family history | Familial disease confirmed at necropsy or surgery | Family history of premature sudden death (<35 yr old) owing to suspected right ventricular dysplasia |
| Familial history (clinical diagnosis based on present criteria) |
* Detected by echocardiography, angiography, MRI, or radionuclide scintigraphy.
Electrocardiogram Evaluation
At least one ECG abnormality (Table 65-3) is detectable in more than 90% of patients with ARVD. A newly proposed marker of delayed right ventricular activation, prolonged S wave upstroke (≥55 ms) in V1 to V3, is the most prevalent ECG marker and correlates with disease severity and ventricular tachycardia induction on electrophysiology testing.17 Signal-averaged ECG is used to detect delayed ventricular activation owing to slowed propagation in the fibrofatty myocardium of ARVD. Late potentials on signal-averaged ECG (two or more of the following: filtered QRS duration ≥114 ms, duration of the low-amplitude signal <40 µV in the terminal portion of filtered QRS (LAS40) ≥38 ms, and root mean square voltage of the terminal 40 ms of filtered QRS (RMS40) <20 µV) form a minor diagnostic criterion for ARVD as per the Task Force criteria. Similar to extent of right precordial T wave inversion, late potentials on signal-averaged ECG are correlated with the extent of right ventricular involvement in ARVD. More recently, our group found that using filtered QRS 110 ms or greater identifies ARVD patients with inducible ventricular tachycardia more accurately than does conventional signal-averaged ECG criteria.18 T wave inversions and signal-averaged ECG abnormalities are more commonly seen in left-sided involvement in ARVD.
TABLE 65-3 Electrocardiogram Features of Arrhythmogenic Right Ventricular Dysplasia‡
* Minor Task Force criterion in the absence of right bundle branch block in individuals >12 yr old.
‡ These electrical abnormalities are believed to be caused by intraventricular myocardial defect (parietal block) rather than definite alteration of conduction in the bundle branch (septal block).
Imaging Techniques and Findings
Chest Radiograph
Chest radiographs generally show cardiac enlargement depending on the stage of the disease, with the cardiothoracic ratio generally less than 0.6. The heart can also show a convexity between the aortic knob and the left ventricle, and there is no pulmonary vascular redistribution.11 Radiographs are not a sensitive modality to diagnose ARVD.
Cine Angiography
Right ventricular cine angiography has been regarded as the traditional gold standard, with high specificity and positive and negative predictive values in ARVD diagnosis. It is performed using two orthogonal views in 30 degrees right anterior oblique and 60 degrees left anterior oblique during sinus rhythm with careful evaluation of structure and function of the right ventricle. Coexistence of subtricuspid and anterior infundibular wall bulging and hypertrophic trabeculae has been shown to have 96% sensitivity and 87.5% specificity in angiographic diagnosis of ARVD.10 Invasiveness of the procedure, frequent ventricular ectopy during catheter manipulation and contrast injection, and interobserver variability in the visual assessment of right ventricular wall motion abnormalities are important limitations. Because of these limitations, and the availability of other, more quantitative imaging tools such as MRI, right ventricular cine angiography plays only a small role today in the evaluation of patients with suspected ARVD. Table 65-4 lists major angiographic features of ARVD.
TABLE 65-4 Angiographic Features of Arrhythmogenic Right Ventricular Dysplasia
Nuclear Ventriculoscintigraphy
Nuclear ventriculoscintigraphy provides information about the size, ejection fraction, contraction pattern of the ventricles, and site of origin of ventricular tachycardia. In most cases of ARVD, the size of the right ventricle is increased, and ejection fraction is diminished. Contraction pattern of the right ventricular wall is asynchronous as a result of the ongoing dysplastic process. Use of specific radionuclide tracers has shown abnormal sympathetic myocardial innervation in ARVD patients, providing a new insight into the arrhythmogenicity of the disease. Myocardial imaging with iodine 123 MIBG and thallium 201 has detected left ventricular involvement early in the course of the disease, suggesting that it may be potentially more sensitive in the diagnosis of ARVD.19,20 Although nuclear imaging can be used in the evaluation of patients with suspected ARVD, it is rarely employed today.
Echocardiography
Two-dimensional echocardiography is an important first-line imaging tool for diagnosis and follow-up in ARVD because of its noninvasiveness, low cost, wide availability, and ease of performance. Echocardiographic abnormalities associated with the Task Force criteria for ARVD are summarized in Table 65-5. A right ventricular outflow tract long-axis diameter greater than 30 mm on echocardiography has been found to have the highest sensitivity and specificity for the diagnosis of ARVD. Right ventricular dysfunction has been defined as a fractional area contraction less than 32%, or the presence of segmental right ventricular wall motion abnormalities (mostly observed in anterior and apical walls) for the purpose of echocardiographic diagnosis of ARVD.21
TABLE 65-5 Echocardiography Features Associated with Task Force Criteria for Arrhythmogenic Right Ventricular Dysplasia
