CHAPTER 19
Thyroid dysfunction
Colin M. Dayan; Onyebuchi E. Okosieme; Peter Taylor
CHAPTER OUTLINE
Biological actions of thyroid hormones
Synthesis, storage and release of thyroid hormones
Iodine and thyroid hormone synthesis
Transport of thyroid hormones in blood
Entry of thyroid hormone into tissues
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Catabolism of thyroid hormones
Nuclear action of thyroid hormones
Control of thyroid hormone synthesis and secretion
Extrathyroidal factors that may affect thyroid function
THE EVALUATION OF THYROID FUNCTION
Clinical evaluation of thyroid status
In vitro tests of thyroid activity and pituitary–thyroid status
Measurement of thyroid stimulating hormone
Free T4 and free T3 measurements
Methods for measuring free thyroid hormones
Validity of commercial methods for free hormone analysis
Nomenclature of free thyroid hormone assays
Selective use of thyroid function tests
Interpreting results of thyroid function tests
Common situations in which TSH results may be misleading
Reference ranges and significant changes
Autoantibodies to thyroidal antigens
Hyperthyroidism or non-thyroidal illness?
Thyroiditis producing hyperthyroidism
Hypothyroidism resulting from Hashimoto thyroiditis
Hypothyroidism and the postpartum period
INTRODUCTION
Thyroid hormones are essential for normal growth, development and metabolism, and their production is tightly regulated through the hypothalamic–pituitary–thyroid axis. Thyroid disease is common, particularly in women, with a prevalence in the community of 3–5%. With the exception of iodine deficiency, which affects millions of people worldwide, diseases that directly affect the thyroid gland are the most common causes of thyroid dysfunction. Pituitary disease and the use of certain drugs that alter thyroid hormone synthesis or metabolism can also give rise to thyroid dysfunction. Any severe illness can produce abnormalities in the results of thyroid function tests that resolve as the patient’s illness improves. Once diagnosed, thyroid disease is usually easily treated, with an excellent long-term outcome for most patients. This chapter outlines thyroid physiology and the pathways of thyroid hormone synthesis and metabolism. The investigations used in diagnosis and management of thyroid disorders are described, together with guidance on their interpretation. The various thyroid disorders are described together with current views on their treatment.
NORMAL THYROID PHYSIOLOGY
The thyroid gland
The thyroid gland is brownish-red in colour and consists of left and right lobes connected by a midline isthmus, typically forming an ‘H’ or ‘U’. It is located anteriorly in the neck deep to the platysma, sternothyroid and sternohyoid muscles. The isthmus lies just below the cricoid cartilage and the lobes extend upward over the lower half of the thyroid cartilage. The pretracheal fascia encloses the thyroid gland and attaches it to the trachea and larynx. This explains why the thyroid gland is seen to move upwards with swallowing.
The thyroid is a highly vascular organ with blood flow ~ 5 mL/min/g of tissue (nearly twice that of the kidneys). It consists of thousands of follicles, each a spheroidal sac of epithelial cells (thyrocytes) surrounding a lumen containing colloid, mainly comprising thyroglobulin (Fig. 19.1). It also contains parafollicular C cells, which secrete calcitonin; calcitonin is discussed in Chapter 6.
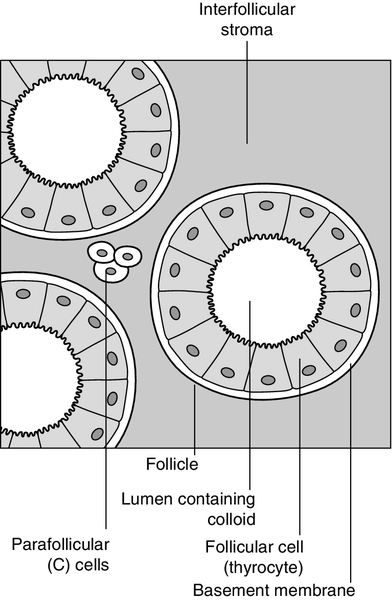
FIGURE 19.1 Structure of the thyroid follicle.
Embryologically, the thyroid is derived from the floor of the pharyngeal cavity and migrates from the base of the tongue to its final position in the neck along the thyroglossal duct. Abnormalities of this migration give rise to ectopic glands that may not function normally. Occasionally, the gland is completely absent.
The primary function of the thyroid is to synthesize and secrete thyroid hormones (Fig. 19.2). Under normal circumstances the gland synthesizes approximately 80 μg (110 nmol) of thyroxine (T4) per day and 6 μg (10 nmol) of 3,5,3′-tri-iodothyronine (T3).
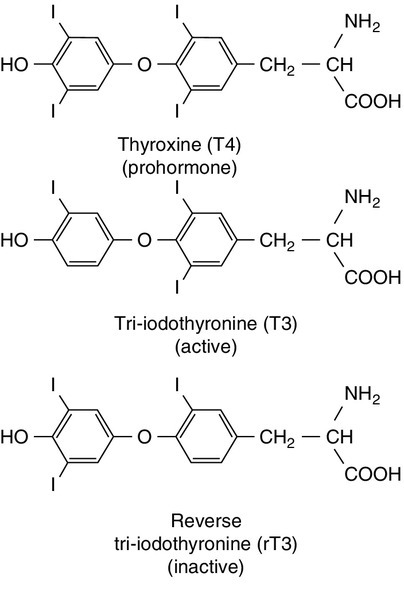
FIGURE 19.2 Structures of the iodothyronines.
Biological actions of thyroid hormones
Thyroid hormones produce enhancement of many intracellular events and promote differentiation and growth; they are essential for normal fetal and neonatal development.
Thyroid hormones increase mitochondrial oxidative phosphorylation and maintain amino acid and electrolyte transport into cells. They increase calorigenesis and oxygen consumption in most tissues, though not in brain. Thyroid hormones stimulate the synthesis of proteins, including structural proteins and enzymes. They regulate all aspects of carbohydrate metabolism, including increasing gluconeogenesis and accelerating insulin degradation. Thyrotoxicosis can, therefore, lead to a deterioration of glycaemic control in diabetes mellitus.
Stimulation of lipid metabolism leads to a fall in plasma cholesterol concentration, as degradation is increased to a greater extent than synthesis. Bone turnover is stimulated, resorption more so than mineralization, raising plasma calcium concentration in some patients. In general, the effect of thyroid hormones on the turnover of any substance will depend on the relative effects on synthesis and degradation. The actions of thyroid hormones increase demand for coenzymes and vitamins.
Complex interactions occur with the sympathetic nervous system and many of the signs and symptoms of hyperthyroidism, including palpitations, tremor and sweating may be attributed to these. For example, thyroid hormones amplify the adrenergic system through the α1 and β3 adrenergic receptors and are also the main determinant of basal metabolic rate (BMR). The increase in metabolic rate involves inhibition of AMP-activated protein kinase (AMPK) in the hypothalamus. However, the mechanisms responsible for many interactions between thyroid hormone and the sympathetic nervous system have not been identified.
Most changes in biochemical parameters produced by changes in thyroid status are not sufficiently specific or of such magnitude to be useful as tests of thyroid function, though the association may be sufficiently close that the finding of an abnormality should provoke a request for testing for previously unsuspected thyroid disease. For example, hypothyroidism is a relatively common and easily treatable cause of raised plasma cholesterol; however, the effect on lipids in subclinical hypothyroidism is modest. Equally, it may be helpful (and avoid unnecessary investigations) to anticipate these changes in cases of known thyroid dysfunction and to expect their normalization after treatment of the thyroid disorder. Some examples are listed in Box 19.1.
Synthesis, storage and release of thyroid hormones
Synthesis of T4 and T3 occurs on thyroglobulin (Tg), a glycoprotein of molecular weight 660 000 Da that contains many tyrosyl residues. Thyroglobulin is synthesized by the thyrocytes and exported to be stored within the colloid of the follicular lumen. Incorporation of iodide into Tg requires hydrogen peroxide and thyroid peroxidase (TPO), an enzyme that is synthesized within the follicular cell and transported to the apical membrane. Thyroid hormones are synthesized at the interface between the apical membrane of the thyrocyte and the colloid of the follicular lumen.
The process of thyroid hormone synthesis, storage and secretion requires a series of highly regulated steps (Fig. 19.3).
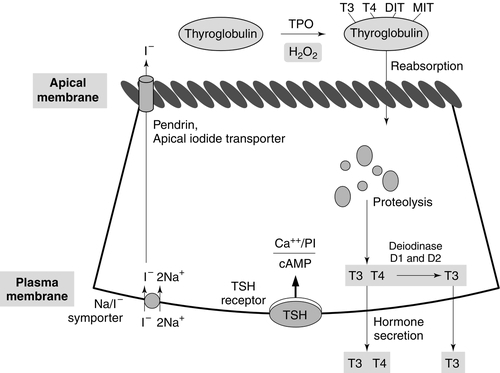
FIGURE 19.3 Sites of biochemical processes within the follicular cell (thyrocyte).
• Oxidation of iodide to iodine by thyroid peroxidase. This occurs on the luminal side of the apical membrane and requires TPO and hydrogen peroxide, which is generated by a calcium-dependent flavoprotein enzyme system situated at the apical membrane.
Recently, the dual oxidase molecules DUOX1 and DUOX2, which contain peroxidase-like and NADPH oxidase-like domains have been identified as essential for H2O2 production. They require maturation or activation factors (DUOXA1 or 2) for translocation of DUOX from the endoplasmic reticulum to the apical plasma membrane where H2O2 production occurs. Mutations in DUOX2 and DUOXA2 have both recently been identified as causes of hypothyroidism, resulting from diminished H2O2 production.
Halogenases collect the iodide removed from thyroglobulin as it is broken down in the cell.
Antithyroid drugs such as propylthiouracil and mercaptoimidazoles (methimazole and carbimazole) exert their action through inhibiting the action of TPO.
• Incorporation of iodine into tyrosyl residues on thyroglobulin. Mono-iodotyrosine and di-iodotyrosine (MIT and DIT) are formed through the actions of TPO (organification).
• Coupling of two iodotyrosyl residues in the thyroglobulin molecule. This is also catalysed by TPO and produces T3 and T4 that remain linked to Tg. When iodine supply is limited, the proportion of T3 produced on Tg increases. Iodinated Tg is stored in the follicular lumen.
• Internalization of Tg and release of T4 and T3. When there is a demand for thyroid hormone, this is signalled by an increase in plasma thyroid stimulating hormone (thyrotrophin; TSH) concentration. Thyroglobulin is then internalized by pinocytosis and appears as colloid droplets that fuse with lysosomes and undergo proteolytic degradation to release T3 and T4. Any MIT and DIT is deiodinated and the iodine conserved.
• Delivery of T4 and T3 into the circulation. This is probably achieved by thyroid hormone transporters. The large stores of T4 and T3 incorporated into thyroglobulin allow secretion of T4 and T3 more quickly when required than if it had it had to be synthesized.
Thyroid stimulating hormone appears to stimulate each of the above processes. It also stimulates the expression of many genes in thyroid tissue including thyroglobulin, sodium iodide symporter (NIS) and interleukin-8. It also causes thyroid hyperplasia and hypertrophy.
Iodine and thyroid hormone synthesis
Iodine deficiency represents a major public health issue for over two billion people who live in areas of iodine deficiency and especially for the 43 million in whom the deficiency is severe enough to cause learning difficulties. The recommended intake of iodine is 150 μg/day for adults but this may rise to 250 μg during pregnancy and lactation. Iodine is obtained from the ingestion of foods such as seafood, seaweed, kelp, dairy products, some vegetables (e.g. soybeans and potatoes) and iodized salt.
High iodine intakes can induce hypothyroidism, goitre and sometimes hyperthyroidism (see later). A large excess of iodide, when given acutely, results in acute inhibition of thyroid hormone release. It also inhibits the adenylate cyclase response to TSH (see below) and iodination of thyroglobulin; this is termed the Wolff–Chaikoff effect. After a few days of exposure to high iodide concentrations, thyroidal iodide uptake is very low, the intrathyroidal iodide concentration falls and the synthesis of iodinated thyroglobulin recommences. This effect can be used to prepare a thyrotoxic patient for thyroidectomy. However, if high iodine intake persists for more than ten days, then thyrotoxicosis can occur in individuals with a pre-existing multinodular goitre or those from iodine deficient areas; this is termed the Jod–Basedow effect.
Transport of thyroid hormones in blood
When T4 and T3 are secreted into the bloodstream, they become bound to plasma carrier proteins. Three proteins share this function: in order of decreasing affinity for iodothyronines they are thyroxine binding globulin (TBG); thyroxine binding prealbumin (TBPA, also known as transthyretin), and albumin. These binding proteins bind approximately 70% (TBG), 20% (TBPA) and 10% (albumin) of the circulating thyroid hormones. T4 is more tightly bound than T3 to each of these proteins. Approximately 0.02% of T4 and 0.2% of T3 are in the unbound or ‘free’ form in plasma in normal subjects, so that, although the total molar concentration of T4 is some 50 times that of T3, the concentration of free T4 is only about three times that of free T3. The half-life of T4 is 5–7 days; that of T3 is 1–2 days.
Free hormone hypothesis
For most of their actions, thyroid hormones need to enter the cell and bind to nuclear receptors. The free hormone hypothesis suggests that only the unbound (free) fraction of thyroid hormone is able to cross the plasma membrane and enter the nucleus. This hypothesis is supported by the observation that, in clinical practice, thyroid status correlates better with the concentration of the free hormone fraction than with the protein-bound fraction. The precise function of TBG and TBPA is unclear, but a number of roles have been suggested including acting as a reserve or buffer to prevent rapid changes in free hormone concentration in blood and also to prevent loss of thyroid hormones through filtration and excretion at the kidney. One further hypothesis is that TBG may be involved in targeting the delivery of thyroid hormone in a differential manner to different organs or facilitating the efflux of thyroid hormones from some tissues. Whatever their function, the absence or overexpression of the transport proteins is not associated with any obvious detrimental effects. There are difficulties in measuring the concentrations of free hormones and this is discussed later in this chapter.
Entry of thyroid hormone into tissues
Specific transporters appear to be required to facilitate the entry of the free thyroid hormones into cells. A number of important thyroid hormone transporters have been characterized. The organic anion transporting polypeptides (OATPs) are one group of important transporters. OATP1C1 appears to be specific for T4 and reverse tri-iodothyronine (3,3′5′-tri-iodothyronine: rT3); it is expressed exclusively in the capillaries of the brain and may be important for T4 transport across the blood–brain barrier. Two L-type amino acid transporters (LAT1 and LAT2) have been identified among the members of the heterodimeric amino acid transporter family, that transport thyroid hormone in many tissues. Human monocarboxylate transporter 8 (MCT8) is another active thyroid hormone transporter. It has a preference for T3 and is expressed in a variety of tissues including the brain, where it appears to be involved in the uptake of T3 by neurons, the primary target for thyroid hormones during brain development. The MCT8 gene is located on the X chromosome and mutations in the gene have been identified in a small number of boys with severe psychomotor retardation and elevated plasma T3 concentration (Allan–Herndon–Dudley syndrome). These mutations result in an inhibition of T3 supply to neurons leading to a defect in brain development. The characteristic pattern of thyroid function tests remains unexplained.
Entry of thyroid hormone into liver cells appears to be ATP-dependent. In rat hepatocytes, it is inhibited by a range of compounds that accumulate in plasma during illness. These compounds include free fatty acids, bilirubin and indoxyl sulphate. The uptake of thyroid hormones by the pituitary appears to occur through a different mechanism from that occurring in the liver. For example, unlike in the liver, the pituitary thyroid hormone transporters are not energy dependent; they can maintain or increase the uptake of T4 and T3 in low energy states and are not inhibited by bilirubin.
Thyroid hormone deiodination and regulation of extrathyroidal T3 production
Thyroxine is produced entirely by the thyroid gland, approximately 80 μg being produced each day. However, only 20% of T3 is produced directly from the thyroid gland; 80% is produced by the extrathyroidal deiodination of T4. Approximately 6 μg of the T3 in blood originates from thyroidal synthesis; 25 μg is formed by 5-deiodination of T4 in extrathyroidal tissues, a reaction catalysed by the D1 and D2 isoenzymes of iodothyronine deiodinase. T4 is considered to be a prohormone, T3 being responsible for the biological actions of thyroid hormones. Most tissues express one or more of a family of three iodothyronine deiodinases (D1, D2 and D3) that are responsible for providing a local source of T3 from T4 or for providing a source of T3 in plasma. In humans, it has proved difficult to determine which deiodinase and which tissues are responsible for providing most of the circulating T3. Classically it has been thought that D1 in liver and kidney provided most circulating T3, but some studies have suggested that, in humans, D2 may also provide an important source. Genetic association studies may provide more conclusive evidence. To date common genetic variants (single nucleotide polymorphisms) in DIO1 have been robustly associated with variation in the T3/T4 ratio in serum at genome wide levels of significance (p = < 1 × 10− 8), but as yet no associations have been observed with single nucleotide polymorphisms in DIO2.
Figure 19.4 summarizes the characteristics of the three deiodinases. All are selenoenzymes requiring adequate intake of dietary selenium for their expression. D1 carries out either 5′-deiodination, giving T3, or 5-deiodination, giving the inactive compound rT3. D2 appears to provide a very important local source of T3 in some tissues, including the brain, pituitary and brown adipose tissue. D3 catalyses the conversion of T4 to rT3 and is thought to be an important extrathyroidal control mechanism to regulate delivery of T3 to its receptors, particularly in the developing fetus.
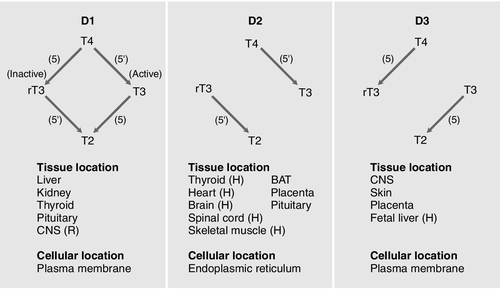
FIGURE 19.4 Characteristics of the iodothyronine deiodinases. In non-thyroidal illness, 5′-mono-deiodination is impaired leading to a decreased production of T3 and impaired breakdown of rT3. BAT, brown adipose tissue; CNS, central nervous system; (H), human not rat; (R), rat not human; T2, 3,3′-di-iodothyronine. (From Beckett G J, Arthur J R. Journal of Endocrinology 2005; 184:455–465. © Society for Endocrinology, with permission.)
Catabolism of thyroid hormones
Sulphation, glucuronidation, deamination, oxidative decarboxylation, ether cleavage and deiodination form the main routes for the inactivation and degradation of thyroid hormone. These metabolites are excreted via bile or in urine.
Nuclear action of thyroid hormones
Tri-iodothyronine exerts its effects through interaction with nuclear thyroid hormone receptors (TR) that have a high affinity and high specificity for T3. Nuclear thyroid hormone receptors are a family of ligand-regulated transcription factors that are associated with chromatin. Several α and β isoforms of TR are produced and these usually dimerize with the retinoid X receptors (RXRs). TRα1 is widely expressed with particularly high expression in cardiac and skeletal muscles, whereas TRβ1 is predominantly expressed in the brain, liver and kidney and TRβ2 is primarily limited to the hypothalamus and the pituitary.
The TR/RXR complex binds to target response elements located in the promoter region of the target genes. The formation of the T3-TR/DNA complex and subsequent recruitment of a variety of transcriptional coactivators leads to activation of the target genes, giving increased mRNA and protein production. In some circumstances, gene expression may be switched off. Mutations in the TRβ gene lead to a decreased affinity of the receptor for T3, leading to the autosomal dominant clinical syndrome of ‘thyroid hormone resistance’ (discussed later in this chapter).
Control of thyroid hormone synthesis and secretion
Classic feedback regulation
The most important regulator of thyroid homoeostasis is TSH. This dimeric peptide hormone comprises a β-subunit, required for binding to the TSH receptor, and an α-subunit. The α-subunit is the same as that of luteinizing hormone, follicle stimulating hormone and human chorionic gonadotrophin. The β-subunit is specific for TSH: both subunits are required for bioactivity. The subunits contain carbohydrate chains that are essential for the biological activity of the molecule. Modifications to these carbohydrate residues occur in different thyroid states, which alter both the bioactivity of the molecule and its plasma half-life. For example, in primary hypothyroidism, TSH has increased bioactivity and an increased plasma retention time, while in secondary hypothyroidism the bioactivity of the molecule is diminished. Most immunoassays are unable to recognize modifications to the carbohydrate chains. There is a circadian rhythm of TSH secretion, plasma concentrations being highest between midnight and 04.00 h and lowest at about midday. Thyroid stimulating hormone release also occurs in a pulsatile fashion. These changes in TSH are not reflected in the serum thyroid hormone concentrations.
The production of TSH is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4 (Fig. 19.5). Free T3 and free T4 inhibit TSH synthesis and release directly, by inhibiting transcription of the TSH subunit genes and indirectly by inhibiting TRH release. Free T3 and free T4 also decrease TSH glycosylation and therefore its biological activity. It is thought that the hypothalamus, via TRH, sets the level of thyroid hormone production required physiologically, and that the pituitary acts as a ‘thyroid-stat’ to maintain the level of thyroid hormone production that has been determined by the hypothalamus. Twin studies have shown that in individuals without thyroid disease, TSH and thyroid hormone concentrations are largely genetically determined. The setpoint in an individual is relatively narrow with intraindividual variation being less than half the interindividual variation.
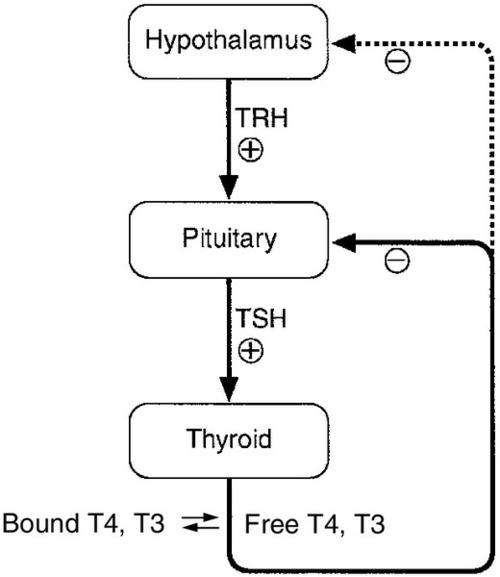
FIGURE 19.5 The ‘classic’ hypothalamo–pituitary–thyroid axis. The production of thyroid stimulating hormone (TSH) is controlled by a stimulatory effect of the hypothalamic tripeptide, thyrotrophin releasing hormone (TRH, thyroliberin), mediated by a negative feedback from circulating free T3 and free T4. Thyroid stimulating hormone may also exert autoregulatory fine control on its own production. Dopamine, somatostatin, glucocorticoids, leptin and catecholamines all may have an influence on the axis.
Other mechanisms
The classic feedback regulation of TSH release described above is an oversimplification. Dopamine, somatostatin and glucocorticoids inhibit TSH release, and these agents, together with cytokines, may be important modifiers of plasma TSH, particularly in patients with non-thyroidal illness (NTI) (Fig. 19.6). Leptin and catecholamines regulate TSH indirectly by stimulating TRH production. There is accumulating evidence that TSH can provide autoregulation via a direct action of TSH on the hypothalamus and at the pituitary. In the pituitary, folliculo-stellate cells have been identified that express TSH receptors: interaction of TSH with these cells may produce fine control on TSH release through the release of cytokines, which in turn inhibit TSH release from thyrotrophs. It has been argued that interaction of TSH receptor antibodies with the folliculo-stellate cells explains why patients with Graves disease may continue to have suppressed TSH weeks or months after normal thyroid hormone concentrations have been achieved through carbimazole therapy.
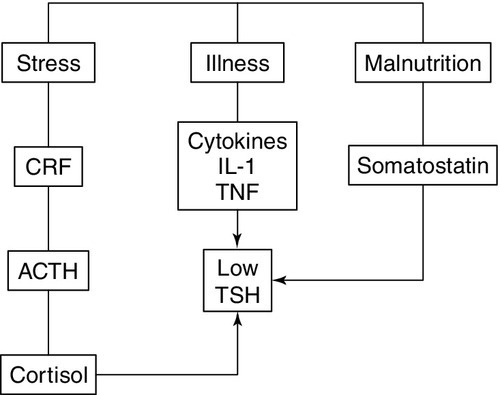
FIGURE 19.6 The hypothalamo–pituitary–thyroid axis in non-thyroidal illness. Dopamine, somatostatin, and glucocorticoids inhibit TSH release, and these agents together with cytokines may be important modifiers of serum TSH, in particular in patients with non-thyroidal illness. ACTH, adrenocorticotrophic hormone; CRF, corticotrophin releasing factor; IL-1, interleukin-1; TNF, tumour necrosis factor.
Thyroid stimulating hormone modifies thyroid hormone synthesis by binding to a specific receptor on the surface of the thyrocyte. The TSH receptor is a single protein with a large extracellular amino-terminal domain involved in the binding of TSH, seven transmembrane domains and a short intracellular carboxy-terminal domain involved in the activation of G-protein modulators of the adenylate cyclase-protein kinase-A system. These characteristics are shared with receptors for gonadotrophins: their extracellular domains show some 40% homology, the remainder being more highly conserved. This may explain the weak thyroid-stimulating activity of human chorionic gonadotrophin (hCG). Binding of TSH results in activation of adenylate cyclase and accumulation of cyclic AMP. The calcium and phosphoinositol signalling pathways may also be activated by TSH. After about an hour, an increase in release of thyroid hormones is seen. In the longer term, TSH increases the synthesis of iodinated thyroglobulin and also causes a general increase in the metabolism, size and activity of the follicular cells.
Extrathyroidal factors that may affect thyroid function
Age
Fetus
Thyroxine and TSH are detectable in fetal plasma at 10–12 weeks of gestation. Plasma concentrations of total and free T4, total and free T3 and TBG increase during gestation, total and free T4 reaching adult concentrations at about 36 weeks, but total and free T3 reaching only the lower limit of the adult range at this time. The concentration of TSH also increases with gestational age but is within or above the adult reference range throughout gestation. Marked time- and organ-specific changes occur throughout gestation in the expression of the deiodinases, which are thought to coordinate the orderly maturation of enzyme systems responsible for development of the brain and the other organ systems in the fetus.
Neonate
During the first 24 h after birth, there is a rapid, transient increase in the release of TSH, T3 and T4 that is considered to be an adaptive response to birth. Thyroid stimulating hormone peaks during the first 30 min, which then stimulates T3 and T4 production during the first 24–36 h of postnatal life. This effect may be attenuated in infants delivered prematurely. Screening for congenital hypothyroidism should be carried out after at least three days to avoid spurious results.
Infancy and childhood
Plasma concentrations of TSH are within the adult range but free T3 is higher than in adults. Free T4 concentrations tend to be at the lower end of the adult range. This hormone pattern suggests it may arise as a result of increased D1 activity in children. After puberty, no major changes in thyroid function occur, except in the pregnant woman.
Elderly
In old age, there is little change in thyroid function tests and age-related reference ranges are not required. There is a modest decrease in T4 secretion but without an accompanying change in circulating T4. A slight fall in T3 may occur in those over 80 years of age. In patients on T4 replacement, the dose of T4 may have to be decreased with age. There is also increasing evidence that the normal TSH distribution curves appear to be shifted towards higher value ranges in individuals aged over 80. Age-specific analysis of TSH concentrations and anti-thyroid antibody titres measured in the National Health and Nutrition Examination Survey (NHANES) demonstrated that 12% of subjects aged 80 and older without any evidence of underlying autoimmune thyroiditis had TSH concentrations > 4.5 mU/L. What is currently unclear is whether higher TSH concentrations confer a survival advantage or simply represent a degree of non-autoimmune age-related thyroid failure.
Pregnancy
In normal pregnancy there is a large increase in plasma TBG concentration that arises from both an oestrogen stimulated increase in synthesis and diminished clearance of the protein. There is also an increase in the pool size of extrathyroidal T4 distribution and an increase in the deiodination of thyroid hormones in the developing placenta. The result of these changes is that, during pregnancy, there is a marked increase in the requirement for iodine and an approximately 50% increase in T4 production occurs if the supply of iodine is adequate.
In order to maintain homoeostasis, an increase in total T4 and total T3 concentrations occurs, which reach a new steady state around mid-gestation. This requirement for increased thyroid hormone production requires an ideal iodine supply during pregnancy of approximately 250 μg/day. Similarly, women with hypothyroidism taking T4 replacement will need an increase in the dose of T4 of approximately 25–50 μg/day during pregnancy within the first six weeks of gestation, aiming for a TSH no higher than 2.5 mU/L. Some centres recommend that women double their pre-pregnancy levothyroxine dose on two days a week as soon as pregnancy is confirmed to reduce the risk of maternal hypothyroidism. Thyroxine replacement should be monitored carefully using both TSH and free T4, since even modest degrees of hypothyroxinaemia in early pregnancy have been associated with an impaired IQ in the infant.
In pregnancy, free T4 and free T3 concentrations may initially show a slight rise, thought to be due to the weak thyroid stimulating action of hCG, present in very high concentration in early pregnancy. This increase in free hormones can lead to suppression of TSH, such that low or sometimes undetectable concentrations may be found in up to 20% of patients during the first trimester. It is important that these changes in early pregnancy are not construed as suggesting that the patient has thyrotoxicosis. As pregnancy progresses, the concentrations of free thyroid hormones fall and TSH begins to rise, although rarely increasing above the reference range seen in non-pregnant subjects. Free thyroid hormone concentration may be lower than those found in non-pregnant women. It is important that trimester-related reference ranges are used for both TSH and free thyroid hormones. Patients can usually return to their pre-pregnancy levothyroxine replacement regimen immediately post-delivery; however, they should be monitored to ensure that this is still the optimal dose.
Hyperemesis gravidarum (severe vomiting in the first trimester of pregnancy) is associated with high plasma total and free thyroid hormone and low TSH concentrations, making it difficult to distinguish this condition from severe true thyrotoxicosis (thyroid crisis). It is believed that hCG is responsible for the thyroid stimulation; the condition usually resolves by the second trimester. Thyroid stimulating hormone receptor antibodies are negative in patients with hyperemesis but positive if the patient has Graves disease.
Thyrotrophin releasing hormone can cross the placenta from mother to fetus, but TSH does not. Thyroid hormones cross the placenta, but this transplacental flux is regulated in part by changes in expression of placental deiodinase as pregnancy progresses. A maternal supply of thyroid hormones (particularly T4) to the fetus is particularly import in the first trimester until a functional fetal thyroid has developed. However, since a fetus with congenital thyroid deficiency develops relatively normally in utero, this suggests that some maternal-fetal transfer of thyroid hormone must be maintained throughout pregnancy. Iodide and antithyroid drugs also pass to the fetus. Indeed, antithyroid drugs are transferred more readily than thyroid hormone, and for this reason treatment of thyrotoxicosis in pregnancy with a ‘block and replace’ regimen (high dose antithyroid drugs ‘rescued’ by simultaneous thyroid hormone replacement) is contraindicated in pregnancy as it would result in a relatively hypothyroid fetus. The treatment of hyperthyroidism in pregnancy requires careful consideration. Radioactive iodine is contraindicated and the choice of antithyroid drug is currently the subject of debate. Propylthiouracil has been traditionally preferred to carbimazole/methimazole owing to fears of scalp and gastro-oesphageal defects in fetuses exposed to carbimazole/methimazole in the first trimester. However, recent concerns have arisen owing to the occurrence of propylthiouracil induced hepatitis, which has occasionally required liver transplantation and rarely has resulted in maternal death. Autoantibodies may cause additional problems in managing patients with hyperthyroidism in pregnancy. In patients with Graves disease, the transplacental passage of thyroid stimulating immunoglobulins may cause transient neonatal thyrotoxicosis, while the presence of anti-TPO antibodies is associated with an increased risk of maternal hypothyroidism and miscarriage.
Non-thyroidal illness
Patients attending or admitted to hospital suffering from any of a wide range of chronic or acute non-thyroidal illnesses (NTI), often have abnormalities in thyroid function tests. A low T3 may often be found even though the patients are clinically euthyroid; this has been termed the sick euthyroid syndrome.
Several mechanisms are involved, including:
• alterations in the hypothalamic–pituitary–thyroid axis leading to decreased hypothalamic stores of TRH and suppression of TSH release due to increased concentrations of dopamine, cytokines, cortisol and somatostatin (see Fig. 19.6). While the degree of TSH suppression in NTI is often not as great as that found in hyperthyroidism, there is considerable overlap in TSH concentrations found in the two conditions
• changes in the affinity characteristics and in the plasma concentrations of the thyroid hormone-binding proteins. These changes give rise to alterations in the plasma concentrations of both the free and total thyroid hormones
• impaired uptake of thyroid hormones by the tissues
• decreased production of T3 in the peripheral tissues
• changes in the T3 occupancy and function of the T3 receptors.
The contribution of each of the above mechanisms may vary with the severity and stage of the illness, and thus the pattern of thyroid function tests may be extremely variable and may mimic the profile seen in either primary or secondary thyroid disease. Interpretation of thyroid function tests is complicated further by the effects of drugs and methodological problems associated with free hormone measurements.
Thyroid hormone metabolism is markedly affected by fasting and illness, with the magnitude of these changes tending to be proportional to the severity of the illness (Fig. 19.7). Extrathyroidal conversion of T4–T3 is reduced, leading to a marked decline in plasma total and free T3, which may fall to undetectable concentrations. Reverse T3 concentration increases, primarily owing to impaired catabolism rather than increased synthesis. These changes are often considered to be an adaptive response for energy conservation, as rT3 is not metabolically active. Concentrations of free fatty acids and other substances that can compete with thyroid hormones for binding to plasma proteins may rise. This can produce a transient increase in free T4. Drugs that compete for T4 binding sites will have a similar effect. Uptake of thyroid hormone into cells may be impaired, either directly by endogenous inhibitors or indirectly as a result of impairment of the active transport systems of the cells. Finally, administration of corticosteroids or dopamine may suppress TSH release.
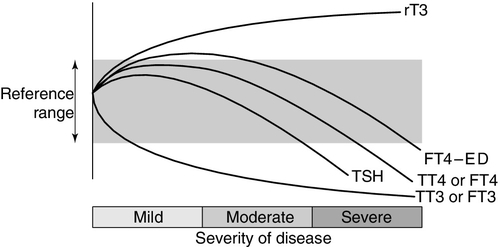
FIGURE 19.7 The effects of illness on the concentration of thyroid hormones and thyrotrophin. The profile for free T4 obtained using equilibrium dialysis is shown (FT4-ED). The concentration of free T4 found using routine assays is method dependent, with some assays showing the profile of FT4-ED, while others follow the profile of total T4 (TT4). Free T3 (FT3) shows a similar profile to total T3 (TT3).
Methodological shortcomings render the results of free hormone analysis liable to serious error. Equilibrium dialysis methods usually show a normal or slightly raised free T4 in patients with illness of mild to moderate severity, but most commercial assays tend to show normal or low free T4 results in sick patients. Normalization or transient rebound changes of thyroid parameters may be seen during recovery from NTI or refeeding after starvation. In particular, TSH may rise transiently into the hypothyroid range. It should be noted that, although TSH is in general the most reliable test of thyroid function, in hospitalized patients a TSH of < 0.1 mU/L is twice as likely to be due to NTI as to hyperthyroidism, and an increased TSH is as likely to be due to recovery from illness as to hypothyroidism. Because of the poor predictive value of thyroid function tests in hospitalized patients, these tests should only be requested if the reason for hospital admission is considered, on clinical grounds, to be related to a thyroid problem. Also, abnormal thyroid function tests during illness may need to be rechecked after the acute illness has resolved.
While the changes in thyroid hormone metabolism associated with illness might theoretically lead to tissue hypothyroidism, most trials of T4 and T3 administration in severe non-thyroidal illness have not shown improved outcome.
Drugs
Drugs may interfere with TSH secretion or the production, secretion, transport and metabolism of thyroid hormones (Table 19.1). Some drugs modify thyroid status while others produce abnormal thyroid function test results in clinically euthyroid subjects. Certain agents (in particular iron preparations) impair the absorption of T4 from the gut, and patients on thyroxine therapy should be advised to take their T4 at least 4 h apart from these medications. Patients should also be advised to avoid caffeine within 45 min of taking levothyroxine as caffeine can also impair absorption. Patients taking levothyroxine are likely to require an increase in replacement dose if drugs such as phenytoin or carbamazepine are also prescribed as they increase hepatic metabolism of T4. Phenytoin, carbamazepine, furosemide and salicylate compete with thyroid hormone binding to plasma binding proteins and may increase plasma free T4 concentrations. The influence of drugs on modifying free thyroid hormone concentrations may be method specific. For example, depending on the method, free T4 may be measured as being normal, high or low in patients given heparin or taking phenytoin or carbamazepine. Amiodarone, lithium and interferon can induce thyroid dysfunction and are covered in detail later in this chapter.
TABLE 19.1
Drugs affecting thyroid function
| Mechanism | Example of drug |
| Decrease in TSH secretion | Dopamine, glucocorticoids, octreotide, cytokines |
| Decrease in thyroid hormone secretion | Lithium, amiodarone, iodide |
| Increase in thyroid hormone secretion | Lithium, amiodarone (rare), iodide |
| Decrease in thyroidal synthesisa | Carbimazole, propylthiouracil, lithium |
| Increase in TBG | Oestrogens, tamoxifen, heroin, methadone, clofibrate, raloxifene |
| Decrease in TBG | Androgens, glucocorticoids, anabolic steroids |
| Displacement of thyroid hormones from plasma proteins | Furosemide, fenclofenac, salicylates, mefenamic acid, carbamazepine |
| Increased hepatic metabolism | Phenytoin, carbamazepine, rifampicin, barbiturates |
| Impaired T4 and T3 conversion | β-antagonists, amiodaronea, radiocontrast dyes, e.g. iopanoic acid |
| Impaired absorption of thyroxineb | Cholestyramine, aluminium hydroxide, ferrous sulphate, calcium salts, sucralfate, soya protein |
| Altered immunityc | Interleukin-1, interferons, tumour necrosis factor-α, interleukin-2 |
Other drugs listed produce abnormal thyroid function tests but patients remain euthyroid. Amiodarone is the exception (see text).
a Cause decrease in thyroid hormone synthesis or secretion and alter thyroid status.
b Interfere with absorption from GI tract. Patients on T4 therapy should be advised to take their T4 at least 4 h apart from these medications.
c These cytokines can cause transient hypothyroidism or thyrotoxicosis, the mechanism of which is unclear.
THE EVALUATION OF THYROID FUNCTION
This section describes the methods that have been developed in order to assess the functional state of the thyroid gland. In most cases, the presenting clinical features will lead to thyroid function tests being performed for confirmation of the provisional diagnosis.
Clinical evaluation of thyroid status
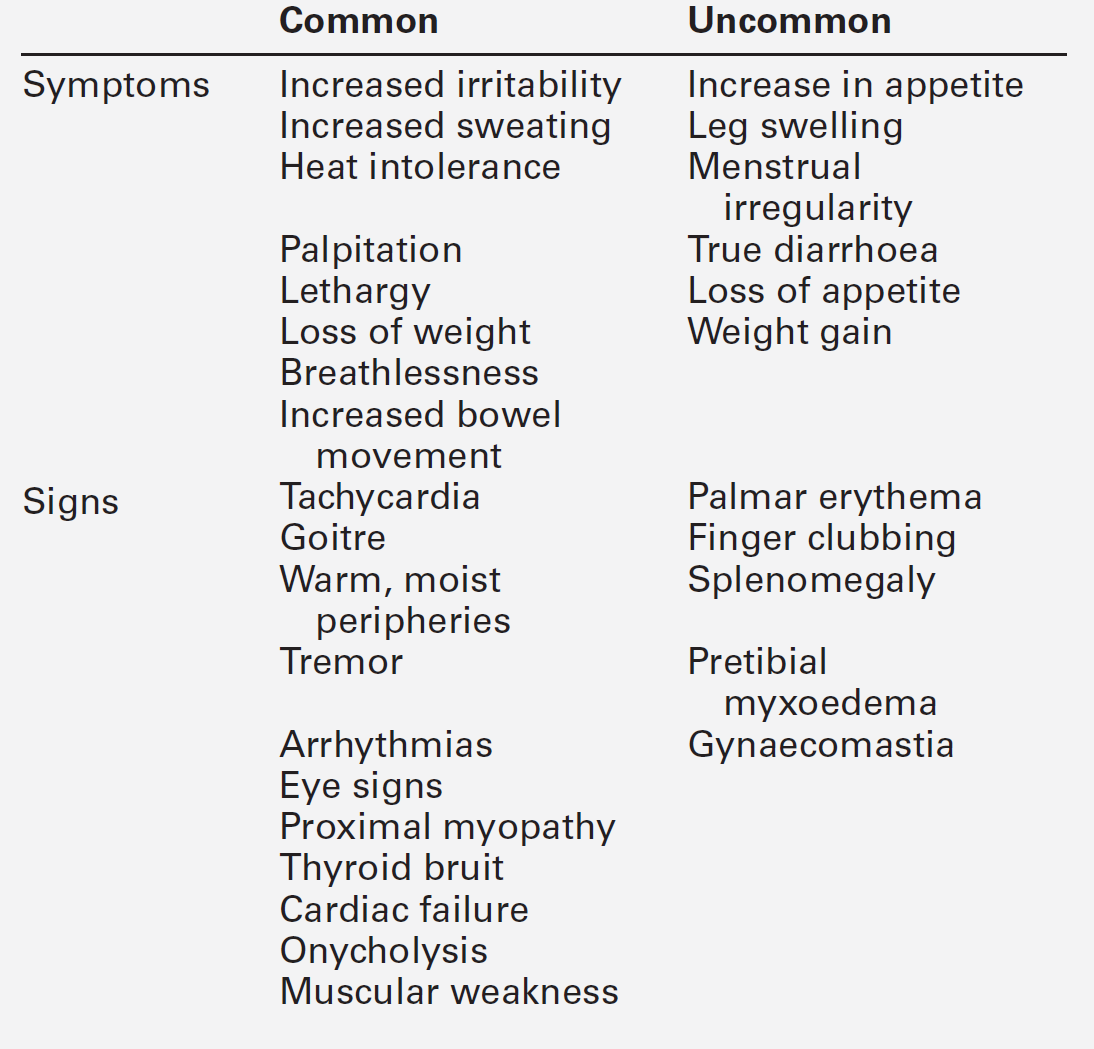
Disordered thyroid function may be detected clinically by demonstrating both changes in the thyroid gland itself and the systemic effects of under- or overproduction of thyroid hormones. In some instances, the clinical presentation may be virtually diagnostic of a specific condition and investigations are required only for confirmation. In others, the clinical features may be less specific and more thorough investigation is required. The signs and symptoms of thyroid dysfunction are listed in Tables 19.3 and 19.6. The normal thyroid can often be palpated and may also be apparent on inspection of the neck, particularly in young, thin women. Abnormal thyroid enlargements may be diffuse or asymmetrical. As a result of connective tissue attachments to the trachea, thyroid swellings move upwards on swallowing and a similar phenomenon is found with tongue protrusion in the case of thyroglossal cysts. The normal gland has a ‘rubbery’ feel; in Graves disease and diffuse colloid goitre, the consistency is softer. By contrast, the thyroid in Hashimoto disease is often rather firm, often with a palpable pyramidal lobe. Thyroid carcinoma (anaplastic) and Riedel thyroiditis result in a rock-hard gland that may, in the case of carcinoma, be irregular in outline. Increased blood flow through a hyperactive gland is often associated with a bruit. This may be maximal over the superior thyroidal artery rather than the gland itself and, in some cases, may also produce a palpable thrill.
In vitro tests of thyroid activity and pituitary–thyroid status
The tests used to investigate thyroid dysfunction can be grouped into:
• tests to elucidate the cause of thyroid dysfunction, e.g. thyroid autoantibody and serum thyroglobulin measurements, thyroid enzyme activities, biopsy of the thyroid, ultrasound and isotopic thyroid scanning
• thyroglobulin measurements, which are used to monitor treatment and detect recurrence in patients with follicular or papillary carcinoma.
Measurement of TSH and thyroid hormones should be performed to determine the patient’s thyroid status before requesting the additional tests that seek to determine the cause of any thyroid dysfunction.
Measurement of thyroid stimulating hormone
The measurement of TSH in a basal serum sample by a sensitive immunometric assay provides the single most sensitive, specific and reliable test of thyroid status in both overt and subclinical primary thyroid disorders. In primary hypothyroidism, TSH is increased, while in primary hyperthyroidism TSH is usually < 0.02 mU/L. However, TSH alone is not a reliable test for detecting thyroid dysfunction arising from hypothalamic–pituitary dysfunction and in other specific instances as outlined below.
Thyroid stimulating hormone is now measured by immunometric assays (IMAs) that utilize non-competitive labelled antibody methods with non-isotopic labels. These IMAs use highly specific monoclonal antibodies raised to epitopes on α- and β-subunits of the TSH molecule. The IMA technique uses a sandwich- assay configuration using two antibodies. The ‘functional sensitivity’ of a TSH assay defines the minimum concentration of TSH that can be quantified in routine use. To obtain the functional sensitivity of a routine assay, a precision profile is constructed using data obtained over many assay runs with multiple batches of reagent and different operators. The concentration of TSH that has a coefficient of variation of 20% taken from the precision profile defines ‘functional sensitivity’. It is essential that TSH assays are sufficiently sensitive for clinical diagnostic purposes and they must have a functional sensitivity of < 0.02 mU/L.
Free T4 and free T3 measurements
Theoretical considerations
The plasma concentrations of free thyroid hormones are extremely low and their quantification in the presence of large concentrations of bound hormones has proved challenging. Poor assay design of many free hormone methods has produced a mass of conflicting literature regarding the typical ranges of free thyroid hormone concentrations in patients with NTI and taking a variety of drugs.
The free hormone concentration results from the equilibrium between the bound (BP) and free hormone.
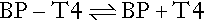
The proportion of T4 that binds to the binding protein is dictated through the law of mass action by the affinity of the binding protein (Keq) multiplied by its concentration. This is known as the ‘relative binding capacity’. Thus, if the affinity or concentration of the binding protein decreases, the proportion of T4 bound will also diminish and the free hormone concentration will increase. In humans, the free T4 fraction is mostly affected by changes in the affinity and concentration of TBG; changes in the affinity and concentration of albumin or transthyretin have little effect on free T4.






