Figure 27-1 A, Homogeneously stained regions and, B,double-minute chromosomes in neuroblastoma.
The clinical stage and age of onset are highly significant independent prognostic variables. For example, a unique presentation of NB, stage IV-S (IV-special) is frequently associated with spontaneous remission. 17 This form of the disease typically presents in infants younger than 1 year of age with evidence of remote disease in the liver and bone marrow, though sparing bone. It is not known whether IV-S NB represents metastatic disease or a multifocal nonclonal disorder of neuroblast development. Stages I and II NB can generally be effectively managed with surgical resection alone, although those rare patients with low-stage disease and adverse biologic markers often require adjuvant chemotherapy. Multimodality therapy, including high-dose chemotherapy, hematopoietic stem cell harvest and rescue, radiation therapy, 123I-MIBG therapy, and Ch14.18 immunotherapy are required to achieve remissions in stage IV (and to a lesser extent stage III) NB, although remission is maintained in less than 40% of patients.
Genetics and Cell Biology
Nonrandom cytogenetic abnormalities are observed in more than 75% of neuroblastomas. The most common of these is deletion or rearrangement of the short arm of chromosome 1, although loss, gain, and rearrangements of chromosomes 10, 11, 14, 17, and 19 have also been reported. Two other unique cytogenetic rearrangements are highly characteristic of neuroblastoma: homogeneous staining regions and double-minute chromosomes (see Figure 27-1). These contain regions of amplification of the N-myc gene, an oncogene with considerable homology to the cellular proto-oncogene c-myc. N-myc amplification is associated with rapid tumor progression, and virtually all neuroblastoma tumor cell lines demonstrate amplified and highly expressed N-myc. 18 Decreased N-myc expression is observed in association with the in vitro differentiation of neuroblastoma cell lines. 19 This observation formed the basis for therapeutic trials demonstrating a survival advantage to patients treated with cis-retinoic acid. 20
Neuroblastoma cells that express the high-affinity nerve growth factor receptor trkA can be terminally differentiated by nerve growth factor and demonstrate morphologic changes typical of ganglionic differentiation. Tumors showing ganglionic differentiation and trkA gene activation have a favorable prognosis. Expression of trkB receptor is associated with poor prognosis tumors and appears to mediate resistance to chemotherapy.
In addition to chromosomal loss on chromosome 1p36, unbalanced loss of heterozygosity at 11q23 is independently associated with decreased event-free survival. Alterations at 11q23 occur in almost one third of neuroblastomas, being most commonly associated with stage IV disease and age at diagnosis greater than 2.5 years. Telomerase expression and telomere length are yet other valuable markers of clinical significance. 21 In particular, short telomere length is predictive of favorable prognosis, regardless of disease stage, whereas long or unchanged telomeres are predictive of poor outcome. Both in vitro and in vivo studies suggest that telomerase inhibition may represent a unique mechanism for novel biological treatment of NB. 22 A small subset of neuroblastomas is inherited in an autosomal dominant fashion. Until recently, the only gene definitively associated with neuroblastoma risk was PHOX2B, also linked to central apnea. 23 Utilizing high-resolution microarray and next-generation sequencing approaches, de novo or inherited missense mutations in the tyrosine kinase domain of the ALK (anaplastic lymphoma kinase) gene on chromosome 2p23 have been observed in many hereditary neuroblastoma families, as well as in sporadic cases, 24 although no clear correlation with stage of disease has been observed. Current Phase I/II clinical trials with ALK inhibitors substantiate the value of such target identification for novel therapies. However, combination whole-exome, genome, and transcriptome sequencing of neuroblastoma identifies few recurrently mutated genes (ALK, PTPN11, ATRX, MYCN, and NRAS) or pathogenic germline variants (ALK, CHEK2, PINK1, and BARD1). 25
Tumors of the Peripheral Nervous System: Ewing Sarcoma and Primitive Neuroectodermal Tumors
Clinical Description and Pathology
The Ewing sarcoma family of tumors (ESFT) make up the second most common bone malignancy after osteosarcoma in children and young adults with a peak incidence at age 15. Rarely, these tumors can arise in the soft tissues. ESFT includes Ewing sarcoma, peripheral primitive neuroectodermal (PPNET), and Askin tumors, among others. These tumors share indistinguishable genetic alterations, immunohistochemical profiles, and lineage-specific marker expression patterns. As in the case of neuroblastoma, therapy for patients with localized PPNET or ES is highly effective, whereas the prognosis for patients with metastatic disease is extremely poor even in the setting of multimodal therapy. Surgery and radiation are used for primary local control with multi agent chemotherapy used to treat systemic disease.
Genetics and Cell Biology
PPNET and ES typically carry a t(11;22)(q24;q12) chromosomal rearrangement, although variant translocations have been observed. 26 The translocation breakpoint has been molecularly cloned and characterized as an in-frame fusion between the 5′ half of the ES gene, EWS, on chromosome 22 and the 3′ half of the human homologue of an ETS transcription family member, FLI1, on chromosome 11. The resultant chimeric protein replaces the DNA-binding domain of EWS with the ETS-like binding domain of Fli-1, retaining the DNA-binding activity of Fli-1. Important transcriptional targets of the EWS-Fli1 transcription factor may include the IGF-I receptor, 27 which is thought to play a role in the pathogenesis of ES. Several studies have indicated the importance of the autocrine stimulation of the insulin-like growth factor I receptor (IGF-IR) for cell transformation and proliferation induced by EWS-Fli1. Small-molecule inhibitors that block the EWS-Fli1 interaction with RNA helicase A showed promise in inhibiting Ewing sarcoma cell growth 28 ; however, weak association of IGF-1R activity in ES cell lines and primary tumors indicate that it is not an ideal druggable target in this tumor. 29 Expression profiling analysis has also revealed that TP53 is transcriptionally upregulated by the EWS-ETS fusion gene. NKX2.2 has been found to be another target gene of EWS-Fli1 that is required for malignant transformation. Several variant translocations have also been identified, invariably fusing the EWS gene to an ETS family member. Interestingly, it has been suggested that the specific fusion protein expressed in ESFT has prognostic significance. 30 In particular, a rearrangement that joins exon 7 of EWS to exon 6 of Fli1 may confer a more favorable outcome. As well, use of RNA sequencing (RNAseq) in EWS-FLI fusion negative ESFTs has identified at least one novel alteration that fuses BCOR (encoding the BCL6 co-repressor) on chromosome X 11p.14 with CCNB3 (encoding the testis-specific cyclin B3) on chromosome X 11p.22, essentially identifying a novel sarcoma phenotype. 31
Rhabdomyosarcoma
Clinical Description and Pathology
Sarcomas arise in supportive tissues that have their origin in embryonic mesenchyme. These tissues include fibrous tissue, muscle, cartilage, and bone. Each of the different sarcomas exhibits evidence of differentiation along one or more of these cellular lineages.
Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood, with approximately 200 new cases annually in the United States, accounting for nearly 10% of all childhood solid tumors. The incidence of RMS is higher in males than in females (1.4:1), and most cases are diagnosed in children under the age of 6. 32 Rhabdomyosarcoma is believed to arise from primitive embryonic mesenchymal cells committed to the skeletal muscle lineage; however, RMS tumors have been found in tissues not usually containing striated muscle, such as the urinary bladder. Multiple histologic subtypes exist, including predominantly embryonal (ERMS; 63% of all tumors) and alveolar (ARMS; 19%) morphologies. ARMS tend to occur in the extremities and exhibit a more aggressive clinical behavior than ERMS, which tend to present in an axial distribution and with a somewhat more favorable prognosis. The management of RMS typically includes local control with both surgery and radiation treatment, with neo-adjuvant chemotherapy being used for management of known and micrometastatic disease. It is notoriously difficult to achieve a sustained remission, or cure, for children with metastatic RMS, particularly those with the alveolar variant. Although the overall survival for childhood RMS is approximately 75%, those with metastatic disease have a less than 20% chance of cure.
Genetics and Cell Biology
Characteristic genetic lesions have been found in both major subtypes of RMS. More than 75% of tumors of the alveolar subtype demonstrate one of two chromosomal translocations, t(2;13)(q35;q14) or t(1;13)(p36;q14), 33,34 which fuse the 5′ DNA-binding region of PAX-3 on chromosome 2 or PAX-7 on chromosome 1, respectively, which are implicated in neuromuscular development, to the 3′ transactivation domain region of the FKHR (FOXO1A) gene—a member of the forkhead family of transcription factors commonly associated with regulation of apoptosis (Figure 27-2 ). Tumors with the t(2;13) translocation have a much poorer prognosis than those with the rarer t(1;3) rearrangement. Interestingly, fusion-negative ARMS tumors are clinically and molecularly indistinguishable from embryonal RMS, and demonstrate outcomes more closely resembling ERMS than fusion-positive ARMS, thus making the presence of the PAX-FKHR fusion a diagnostic criterion for ARMS. 35 Additional epigenetic or genetic events seem required for RMS tumorigenesis. PAX-3-FKHR fusion is associated with increased expression of c-met. Met is the receptor tyrosine kinase for hepatocyte growth factor/scatter factor and is overexpressed in embryonal and alveolar RMS. Other frequently reported genetic alterations that may be common to embryonal and alveolar RMS include activated forms of N- and K-RAS, inactivating TP53 mutations, and amplification and overexpression of MDM2, CDK-4, and N-MYC.
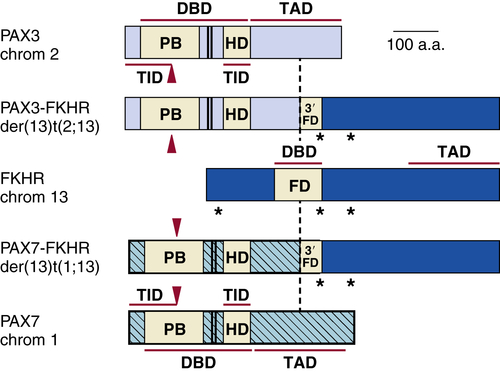
Figure 27-2 Translocation breakpoints in rhabdomyosarcoma.
At the molecular level, embryonal tumors are characterized by loss of heterozygosity (LOH) at the 11p15 locus, which is of particular interest because this region harbors the IGF2 gene. 36 The LOH at 11p15 occurs by loss of maternal and duplication of paternal chromosomal material. 37 Although LOH is normally associated with loss of tumor suppressor gene activity, in this instance LOH with paternal duplication may result in activation of IGF2. This occurs because IGF2 is now known to be normally imprinted—that is, this gene is normally transcriptionally silent at the maternal allele, with only the paternal allele being transcriptionally active. Thus, LOH with paternal duplication potentially leads to a twofold gene-dosage effect of the IGF2 locus.
In addition to the somatic molecular changes associated with RMS, the tumor is also observed in hereditary cancer syndromes, including Li-Fraumeni syndrome (see following section), in which carriers harbor constitutional mutations of the TP53 tumor suppressor gene. The possible importance of the patched gene, PTCH, in the development of RMS is suggested by the finding that mice lacking this gene develop RMS. PTCH is mutated in the germline of patients with Gorlin syndrome, a disorder that includes predisposition to tumor (medulloblastoma) development. Strikingly, PTCH is shown to regulate another gene, GLI, which is found to be amplified in RMS and Gorlin syndrome–associated tumors. Activation of the HRAS oncogene by heterozygous germline mutations predisposes to RMS in Costello syndrome, 38 further highlighting the multiple molecular pathways associated with rhabdomyosarcomagenesis.
Childhood Sarcomas: Osteosarcoma
Clinical Description and Pathology
Osteosarcoma (OS) occurs most frequently in adolescence during a period of rapid bone growth. It is the most common tumor in this age group other than those of hematopoietic tissues. OS most commonly occurs at metaphyseal growth plates of long bones, develops earlier in girls than in boys, and is more frequent in taller children. These observations suggest an important role for cellular proliferation in the oncogenic conversion of immature bone precursors from which these tumors are thought to arise. The histologic diagnosis of OS is made when tumor osteoid and disorganized bone can be identified within malignant stromal tissues. A wide range of histologic patterns is seen, although the natural history of these variants is not yet clinically distinguishable. Tumors are classified as osteoblastic, chondroblastic, or fibroblastic OS depending on whether the predominant differentiation is a long bone, cartilage, or stromal tissue pathway, respectively. Surgery is the primary therapeutic modality in the management of osteosarcoma. Neo-adjuvant chemotherapy is used to control micrometastases, which are present in 75% of patients. The response to chemotherapy, as measured by histologic grading of the degree of tumor necrosis, is a key prognostic factor. Biologic response modifiers, monoclonal antibodies, and targeted small-molecule kinase inhibitors have had no impact on the treatment of osteosarcoma.
Genetics and Cell Biology
OS is characterized by the presence of complex unbalanced karyotypes. 39 Combined inactivation of the RB1 and TP53 tumor suppressor pathways are observed in most OS, indicating important roles for both these genes in OS pathogenesis. Further evidence for the role of p53 in OS pathogenesis includes the predisposition of patients with germline TP53 mutations (Li-Fraumeni syndrome [LFS]) to develop OS. There is low prognostic significance of TP53 mutations in sporadic OS, with no impact on distant recurrence. Furthermore, p53 status is concordant in paired samples of primary and distant metastases, suggesting that p53 pathway alterations may occur early in OS pathogenesis. Modifying effects of other expressed genes are being explored in OS. In particular, amplification of chromosome 12q13 region (containing MDM2 and CDK4) or deletion of INK4A can disrupt both the p53 and RB pathways. Many recurrent, nonrandom chromosomal abnormalities are observed in OS. Common numerical abnormalities in OS include gain of chromosome 1; loss of chromosomes 9, 10, 13 (at the RB1 locus), and/or 17 (at the TPS3 locus); and partial or complete loss of the long arm of chromosome 6. Frequent structural abnormalities include rearrangements of chromosomes 11, 19, and 20. Genome-wide efforts to identify potential tumor suppressor genes associated with LOH in OS have identified at least one novel locus encompassing the LSAMP gene on chromosome 3p13.4. 40 Examination of 38 chromosomal arms from OS tumor samples for LOH has found that the mean frequency of LOH is 30.79% for any chromosome arm, an unusually high mean frequency for a childhood tumor. Moreover, several chromosome arms (3q, 13q, 17p, and 18q) underwent LOH with a frequency more than two standard deviations higher than the average (p < 0.002). 26 Further mitotic mapping has identified minimal regions thought to contain candidate tumor suppressor loci on chromosomal arms 3q26.2 and 18q21.33, 41 though specific gene identification has been elusive. Finally, an intriguing mechanism of chromosomal instability defined as “chromothripsis,” in which chromosomal fragments undergo a catastrophic one-time rearrangement, has been observed in about 25% of osteosarcomas—far greater than the 2% to 3% observed in other human cancers. 42
Abnormalities of bone growth and remodeling are thought to play an important role in the pathogenesis of OS. Normal bone repair involves proliferation of mesenchymally derived precursor cells mediated by platelet-derived growth factor (PDGF), epidermal growth factor (EGF), IGFs, interferon-α, and other mitogens. The PDGF receptor encodes a tyrosine kinase; when PDGF binds to its receptor, it induces expression of fos, myc, and a cascade of cellular genes important for initiating proliferation. EGF may also play a role in OS pathogenesis. Some OS cell lines express EGF receptor and proliferate in response to exposure to EGF. The mitogenic response to both EGF and PDGF may be blocked by TGF-β, which at low doses may stimulate the proliferation of cells.
Interest in other signaling pathways has implicated the Fas cell death pathway in determining chemosensitivity and metastatic behavior in OS. 43 Tumor cells expressing surface Fas will apoptose when Fas ligand (FasL) is present unless a mechanism of resistance is present. This has been suggested by studies in which metastasis-prone OS cell lines that have been transfected with Fas demonstrate reduced metastatic potential. In addition, overexpression of CyclinE1, which promotes oncogenic transformation of osteoblasts and confers resistance to cisplatin (a drug that forms the backbone of osteosarcoma therapy), offers new avenues for therapy. 44
Cancer Predisposition Syndromes
Several hereditary cancer syndromes are associated with the occurrence of childhood as well as adult-onset neoplasms. Although it is beyond the scope of this chapter to describe them all, it is worthwhile to discuss a few to highlight the important molecular basis on which these disorders develop (Table 27-2 ).
Li-Fraumeni Syndrome
Li-Fraumeni familial cancer syndrome is the prototypical familial cancer predisposition syndrome. The definition of classical LFS requires a proband with a sarcoma diagnosed before 45 years of age, a first-degree relative diagnosed as having any cancer when younger than 45 years, and a first- or second degree relative with a diagnosis of cancer when younger than 45 years or a sarcoma at any age. 45 The classic spectrum of tumors that includes soft tissue sarcomas, osteosarcomas, breast cancer, brain tumors, leukemia, and adrenocortical carcinoma (ACC) has been overwhelmingly substantiated by numerous subsequent studies, although other cancers, usually of particularly early age of onset, are also observed. Similar patterns of cancer that do not meet the classic definition have been termed Li-Fraumeni–like syndrome (LFS-L). The sensitivity and specificity of the Chompret criteria are 82% and 58%, respectively, making it perhaps the most rigorous and relevant definition to justify TP53 mutation analysis. 46
Germline alterations of the TP53 tumor suppressor gene are associated with LFS. 47,48 These are primarily missense mutations that yield a stabilized mutant protein. The spectrum of germline TP53 mutations is similar to that of somatic mutations found in a wide variety of tumors. Carriers are heterozygous for the mutation, and in tumors derived from these individuals, the second (wild-type) allele is frequently deleted or mutated, leading to functional inactivation. Several comprehensive databases document all reported germline (and somatic) TP53 mutations and are of particular value in evaluating novel mutations as well as phenotype-genotype correlations. Approximately 75% of “classic” LFS families have detectable TP53 alterations. It is not clear whether the remainder are associated with the presence of modifier genes, promoter defects yielding abnormalities of p53 expression, or simply the result of weak phenotype-genotype correlations (i.e., the broad clinical definition encompasses families that are not actual members of LFS). The variability in type of cancer and age of onset within and between LFS families suggests that expression of modifier genes might influence the underlying mutant TP53 genotype. Several of these have been described, including those that accelerate age of tumor onset in TP53 mutation carriers such as a single-nucleotide polymorphism (SNP) in the promoter of the MDM2 gene (SNP 309) that is involved in the p53 degradation pathway 49 ; accelerated telomere attrition—perhaps inducing chromosomal chromothripsis in somatic cells 50 ; and increased constitutional copy number variation (CNVs). 51 Others, such as a 16-bp duplication in TP53 intron 3 (PIN3), delay tumor onset by up to 19 years. 52 Until recently, options for intervention in LFS were thought to be limited, but two studies have clearly demonstrated the value of total body imaging (with or without biochemical marker studies) in TP53 mutation carriers (Table 27-3 ). 53,54 This has proven to be effective in early tumor detection, which leads to improved survival—offering hope for these patients that the combination of molecular testing with early clinical surveillance can interfere with the natural history of the disease.
Hereditary Paraganglioma Syndromes
Paragangliomas are benign non–catecholamine-secreting tumors that often occur in the head and neck region, along the parasympathetic chain. Catecholamine-secreting tumors can develop along the sympathetic chain, in the adrenal medulla (pheochromocytoma) alongside the aortopulmonary vasculature, in the organ of Zuckerkandl, or even in the bladder and vas deferens. Paragangliomas have an estimated population incidence of 1 in 30,000. However, in the presence of an underlying germline succinyl dehydrogenase (SDHx) mutation, the tumor rate is extraordinarily high, with disease penetrance approaching 80%. 55,56 Germline SHDx mutations are identified in nearly 30% of patients with nonmetastatic paragangliomas and pheochromocytomas and in 44% of adults and 81% of pediatric patients with metastatic disease. 57 Other tumors, including renal cell carcinoma, oncocytoma, papillary thyroid cancer, pituitary tumors, gastrointestinal stromal tumor (GIST), and even neuroblastoma, are observed in SDHx mutation carriers. Although patients with localized asymptomatic disease are frequently observed, those with metastatic disease are particularly difficult to treat, often requiring multiple surgical procedures. Systemic chemotherapy is generally not effective, although the introduction of multitargeted kinase inhibitors provides a new avenue of molecularly targeted therapy. 58
Table 27-2
Hereditary Syndromes Associated with Childhood Neoplasms
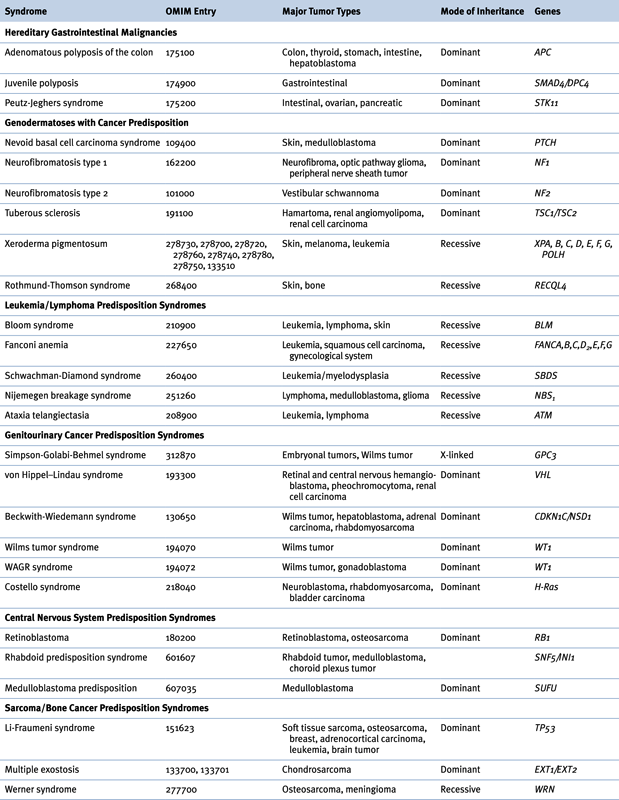
OMIM, Online Mendelian Inheritance in Man.
Table 27-3
Clinical Criteria for Classic Li-Fraumeni Syndrome, LFS-Like Criteria, and Chompret Criteria

Genetics
Succinate dehydrogenase (SDH) is part of respiratory complex II in the mitochondrion, and this enzyme complex is responsible for converting succinate to fumarate as part of the Krebs cycle. SDH is composed of four distinct proteins called SDHA, SDHB, SDHC, and SDHD. 59 A fifth gene called SDHAF2, or SDH Assembly Factor 2, is responsible for assembling all of the individual SDH proteins into a fully functioning enzyme complex. 60 Germline mutations in each of these SDHx genes may lead to development of paragangliomas or pheochromocytomas. Lack of a functioning SDH complex leads to increased succinate, with subsequent increases in HIF signaling and possible histone deregulation. Germline mutations in other genes such as NF1, VHL, RET, TMEM127, and MAX also have been associated with the development of paragangliomas and pheochromocytomas (Figure 27-3 ). Based on gene expression and pathway analysis, these tumors can be divided into two different clusters that correspond to their underlying gene mutations: Cluster 1 (Cluster 1A: SDHx, Cluster 1B: VHL) associated with pseudohypoxia and aberrant VEGF signaling, and Cluster 2 (RET/NF1/TMEM127/MAX) associated with aberrant kinase signaling pathways.
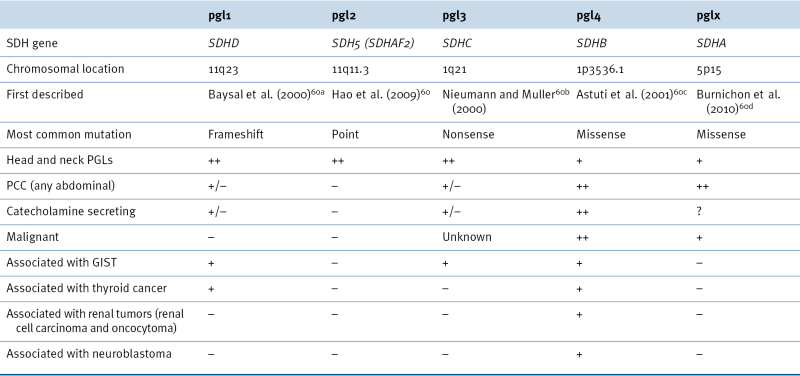
The phenotype associated with each SDHx gene mutation leads to a different disease phenotype and clinical presentation, as outlined in Table 27-4 . 59 To facilitate genetic diagnosis, risk assessment and treatment options, it is now possible to test for all the SDHx genes simultaneously.
Regular surveillance can detect early tumors in patients with underlying germline SDHx mutations. As with surveillance in TP53 mutation carriers, this is important so that smaller, asymptomatic SDH-deficient tumors can be removed before they transform to malignant and metastatic disease. Although no formalized screening guidelines exist, many clinicians will perform annual physical examinations, blood pressure checks (for hypertension due to increased catecholamines), and blood work for serum metabolites. Previously, urine catecholamines were examined from 24-hour urine specimens, but many clinicians have eliminated urine screening in favor of serum testing. Fractionated plasma metanephrines are the most sensitive and specific serum test for detecting secreting paragangliomas and pheochromocytomas. 61 Increased methoxytyramine, a metabolite of dopamine, seems to be helpful for predicting the likelihood of metastatic disease and for distinguishing SDH-related tumors from VHL-related tumors. However, testing of methoxytyramine remains difficult to obtain on a clinical basis.
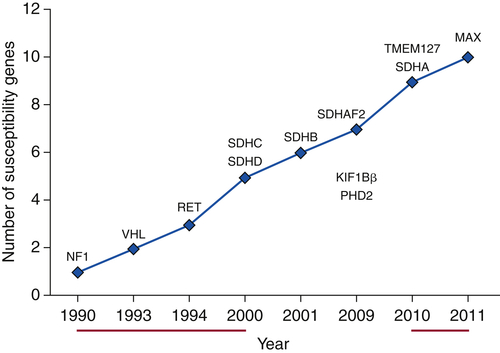
Figure 27-3 Accelerated discovery of genes associated with predisposition to hereditary pheochromocytoma/paraganglioma syndromes.
Regular imaging has been demonstrated by several groups to be very effective at identifying SDH-related tumors, especially in the setting of negative biochemical results. 62 Screening approaches using rapid-sequence whole-body magnetic resonance imaging (MRI) in conjunction with urinary and or fractionated plasma metanephrine levels are being used widely. Abnormal MRI results (or biochemical results) are followed with positron emission tomography (PET) imaging to refine the anatomical location of the tumor.
Beckwith-Wiedemann Syndrome
Beckwith-Wiedemann syndrome occurs with a frequency of 1 in 13,700 births. BWS is associated with a wide spectrum of phenotypic stigmata, including hemihypertrophy/hemihyperplasia, exomphalos, macroglossia, gigantism, and ear pits (posterior aspect of the pinna). Laboratory findings may include profound neonatal hypoglycemia, polycythemia, hypocalcemia, hypertriglyceridemia, hypercholesterolemia, and high serum α-fetoprotein (AFP) level. With increasing age, phenotypic and laboratory features of BWS become less pronounced. Although neurocognitive defects are not universal in BWS, early diagnosis of the condition is crucial to avoid deleterious neurologic effects of neonatal hypoglycemia and to initiate an appropriate screening protocol for tumor development. The increased risk for tumor formation in BWS patients is estimated at 7.5% and is further increased to 10% if hemihyperplasia is present. Tumors occurring with the highest frequency include Wilms tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, and ACC.
Table 27-4
SDHX Genotype: Phenotype Correlations
The genetic basis of BWS is complex, and it does not appear that a single gene is responsible for the BWS phenotype. Various 11p15 chromosomal or molecular alterations have been associated with the BWS phenotype (Table 27-5 ) and its tumors. 63 Abnormalities in this region affect an imprinted domain, indicating that it is more likely that normal gene regulation in this part of chromosome 11p15 occurs in a regional manner and may depend on various interdependent factors or genes. These include the paternally expressed genes IGF2 and KCNQ10T1 and the maternally expressed genes H19, CDKN1C, and KCNQ1. Paternal uniparental disomy, in which two alleles are inherited from one parent (the father), has been reported in approximately 15% of sporadic BWS patients. 64 The insulin/IGF2 region is always represented in the uniparental disomy, although the extent of chromosomal involvement is highly variable. Alterations in allele-specific DNA methylation of IGF2 and H19 reflect this paternal imprinting phenomenon. 64 As with other cancer susceptibility syndromes, effective clinical surveillance protocols regularly identify tumors with demonstrable improved outcomes. Regular (every 3 months) AFP levels and abdominal/pelvic ultrasound are recommended until the affected child is about 9 years old and generally beyond the risk age for the associated tumors.
Gorlin Syndrome
Nevoid basal cell carcinoma syndrome, or Gorlin syndrome, is a rare autosomal dominant disorder characterized by multiple basal cell carcinomas, developmental defects including bifid ribs and other spine and rib abnormalities, palmar and plantar pits, odontogenic keratocysts, and generalized overgrowth. 65 The sonic hedgehog (SHH) signaling pathway directs embryonic development of a spectrum of organisms. Gorlin syndrome appears to be caused by germline mutations of the tumor suppressor gene PTCH, a receptor for SHH. Medulloblastoma develops in approximately 5% of patients with Gorlin syndrome. Furthermore, approximately 10% of patients diagnosed with medulloblastoma by the age of 2 years are found to have other phenotypic features consistent with Gorlin syndrome and harbor germline PTCH mutations. 66 Although Gorlin syndrome develops in individuals with germline mutations of PTCH, a subset of children with medulloblastoma harbor germline mutations of another gene, SUFU, in the SHH pathway, with accompanying LOH in the tumors.
Multiple Endocrine Neoplasia
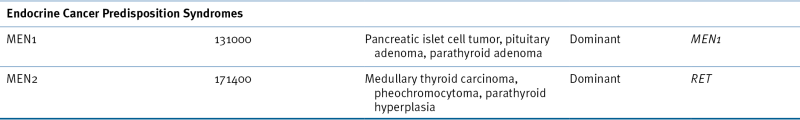
The multiple endocrine neoplasia (MEN) disorders comprise at least three different diseases—MEN type 1, MEN type 2A, and MEN type 2B, which are all cancer predisposition syndromes that affect different endocrine organs. The most common features of MEN type 1 are parathyroid adenomas (about 90% of cases), pancreatic islet cell tumors (50% to 75% of cases), and pituitary adenomas (25% to 65% of cases). 67 MEN2A is associated with medullary thyroid carcinoma, parathyroid adenoma, and pheochromocytoma. MEN2B is a related disorder, but with onset of the tumors in early infancy, ganglioneuroma of the gastrointestinal tract, and skeletal abnormalities.
Although MEN1 is caused by mutation in the tumor-suppressor gene, MEN1, MEN2A, and MEN2B are caused by mutations in the proto-oncogene RET. Further studies confirmed constitutional mutations in the RET
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading. Learn more here






