Mechanisms of Thyroid Hormone Action
Synthesis and Control of Secretion of Thyroid Hormones
Thyroid Hormone Action in the Periphery
Accumulation of Thyroid Hormone in Cells
Deiodinases in Thyroid Hormone Activation and Inactivation
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
History of the Mechanism of Action of Thyroid Hormones
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
Discovery That Thyroid Hormones Regulate Specific mRNAs and Gene Transcription
Discovery That TRs Bind Preferentially to Active Chromatin and Specific DNA Sequences
Thyroid Hormone Receptors: Mechanism
Receptor Structure and Function
Hormone Effects on Receptor Structure
LBD H12 Position and Coregulator Interactions
TR Antagonists: The Extension Hypothesis
Other Influences of H12 Rearrangement
Ligand Stabilizes LBD Structure
Structure and Function of TR Isoforms
TR Mutations in Disease: Structure-Function
Gene and Context-Specific Variations in TR Actions
The award of the 1909 Nobel Prize to Theodore Kocher for pioneering work on thyroid gland surgery lent timely recognition to the field of the thyroid gland and its actions, an area of study that was already several centuries in the making.1 Several Renaissance scholars, including Leonardo da Vinci, contributed to the early description of the thyroid gland and the realization that its swelling led to goiter. Later investigators linked goiter and cretinism to dietary deficiency of iodine (a key component of thyroid hormone) and uncovered links between altered thyroid gland function and apparently unconnected symptoms of hyperthyroidism (thyrotoxicosis or Graves’ disease) and hypothyroidism (or myxedema). Classic experiments of Murray in 1891 revealed that sheep thyroid gland extracts could be used to treat, and even cure, myxedema. Later searches for the active principle culminated in the isolation of thyroxine (T4; 3,5,3′,5′-tetra-iodo-l-thyronine) in 1914 by Kendall (who later, with others, received the Nobel Prize for discovering the therapeutic effects of the adrenal steroid cortisone on rheumatoid arthritis), the description of thyroxine structure and chemical synthesis by Harrington in the 1920s and the discovery of the major active form of thyroid hormone, triiodothyronine (T3; 3,5,3′-tri-iodo-l-thyronine), by Pitt-Rivers about 3 decades later. It is now known that thyroid hormones influence virtually every tissue and cell type; they play important roles in growth, development, and differentiation in fetal life and early childhood and act as master regulators of multiple metabolic processes in the adult. The past 50 years have witnessed great increases in our knowledge of thyroid hormone action, with the last 2 decades in particular producing very rapid advancements in understanding the functions of thyroid hormone receptors (TRs), members of the nuclear hormone receptor (NR) family. In this chapter, we review current knowledge of the mechanisms of thyroid hormone action.2–5
Synthesis and Control of Secretion of Thyroid Hormones
1. Spontaneous release of hormones from the hypothalamus and pituitary that stimulate thyroid hormone production
2. Feedback inhibition of production and release of these hypothalamic and pituitary hormones
3. Rates of intracellular conversion of T4 to T3
4. Metabolic destruction of both of these forms of the hormone6
Thyroid hormones are derivatives of tyrosine that are produced in the thyroid gland and secreted into the general circulation. Thyroid hormone synthesis occurs via iodination of tyrosine residues in a large protein called thyroglobulin, coupling of iodinated tyrosines, and enzymatic cleavage to liberate free thyroxine (T4). Recent human genetic linkage studies suggest that regulation of thyroid hormone production is complex, with up to eight different loci influencing biological set-point.7–9 Some of these probably correspond to known genes involved in synthesis of thyroid hormones and feedback inhibition of thyroid hormone production (see later discussion), but most of these loci are not identified.
T4 is the major form of thyroid hormone secreted from the gland, and much of this T4 is converted to T3 in peripheral tissues. Since T3 binds to TRs with higher affinity than T4, it is thought T3 is the major active form of thyroid hormone. Smaller amounts of alternate forms of thyroid hormone are also made in, and secreted by, the thyroid gland. These products are metabolites of thyroxine and include T3, reverse T3 (rT3), and others. Overall amounts of secreted T4 are much higher (more than 20-fold) than any of these alternate forms of thyroid hormone. The thyroid gland may also produce thyronamines (discussed in more detail later10), compounds that may constitute part of a completely distinct arm of the thyroid hormone signaling system; circulating levels of the most potent currently known thyronamine (3-iodothyronamine) appear higher than those of T3.
The negative feedback loop that controls thyroid hormone levels works through the hypothalamus and pituitary (hypothalamic-pituitary-thyroid [HPT] axis (Fig. 5-1). Thyrotropin-releasing hormone (TRH) is released from the hypothalamus and stimulates the synthesis and release of thyroid-stimulating hormone (TSH) by thyrotrophic cells in the pituitary gland. TSH is released into the circulation, where it stimulates the synthesis and release of T4 and to a lesser extent, T3. The circulating thyroid hormones block release of both TRH and TSH through repression of the transcription of genes that encode the TRH pre-pro-hormone and the α and β subunits of TSH and also by down-regulating TRH receptors in the pituitary. Together, these actions result in repression of hormone synthesis and release in response to high circulating levels of T4 and T3. This process helps maintain thyroid hormone levels within defined limits.
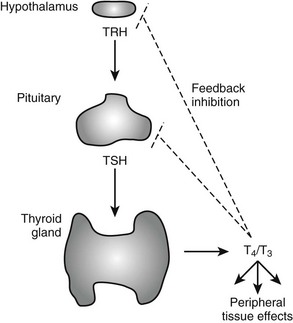
FIGURE 5-1 Regulation of thyroid hormone production in the hypothalamic-pituitary axis. Thyroid hormone is produced in the thyroid gland under control of thyroid-stimulating hormone (TSH). Circulating hormones exert multiple effects on different tissues in the periphery and also feed back to inhibit hypothalamic signals that stimulate TSH release and pituitary production of TSH, thereby maintaining thyroid hormone levels in defined limits.
Thyroid Hormone Action in the Periphery
Both major forms of thyroid hormone (T4 and T3) are transported in the circulation in complex with plasma proteins (Fig. 5-2). The ratio of total T4 to T3 in plasma is about 60:1. This is higher than the 20-fold ratio of T4 to T3 that is initially secreted by the thyroid gland because of greater plasma binding of T4 versus T3, resulting in greater clearance rates of T3. About 99.98% of total circulating T4 and 99.7% of T3 form noncovalent interactions with serum proteins, mostly to thyroxine-binding globulin but to a lesser extent to thyroxine-binding prealbumin, albumin and lipoproteins. The fact that less T3 circulates in complex with plasma proteins means that the ratio of free T4 to T3 is around three- to sixfold, with typical circulating free hormone levels around 20 picomolar (pM) T4 and 6 pM T3, respectively. The free fraction is biologically active and can enter target tissues. Thus, interactions with plasma binding proteins help to ensure even hormone delivery throughout the body.
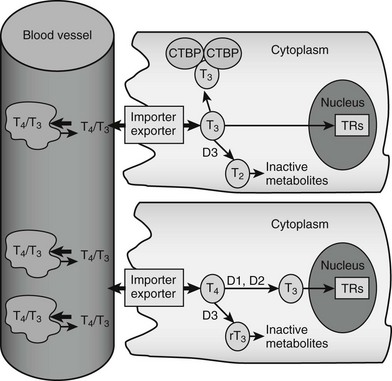
FIGURE 5-2 Peripheral actions of thyroid hormone. The figure summarizes possible fates of thyroid hormone in the periphery. Hormones are mostly transported as complexes with serum binding globulins with limited amounts of free T4 and T3. Hormones are actively taken up into cells by facilitated transport mechanisms, where they undergo several different fates. The upper cell represents possible intracellular fates of T3; it can be sequestered in complexes with CTBP dimers, undergo conversion to inactive metabolites, or enter the nucleus to interact with TRs. The lower cell represents fates of T4, which is either activated by metabolic modification to form T3 or converted to inactive metabolites. It should be noted that expression of D1 and D2 is under transcriptional control, making this step an important control point in thyroid hormone response.
Import and Export
Thyroid hormone entry into cells is mediated by specific transporters (see Fig. 5-2).11–13 While early models suggested that lipophilic thyroid hormone molecules enter cells by diffusion across the plasma membrane, and this may occur to some extent, it is now clear that T4 and T3 entry mostly involves facilitated transport, a form of passive diffusion that requires membrane transport proteins.
Accumulation of Thyroid Hormone in Cells
T3 binds with nanomolar affinity to an intracellular protein that was originally called cytoplasmic T3-binding protein (CTBP).14 This protein is found at high abundance in the kidney and, to a lesser extent, in liver. Because CTBP was later found to be homologous to mu-crystallin, a protein abundant in the lens of the kangaroo eye, the gene is now often called CRYM. Overexpression of CRYM in stable cell lines increases maximal cellular T3 binding capacity and decreases T3 efflux rates. However, these effects are coupled to reduced transcriptional responses to T3, suggesting that CRYM sequesters T3 in an inactive cytoplasmic complex, away from nuclear TRs.
CRYM is important for thyroid hormone action in vivo. Mice with a targeted CRYM gene deletion appear normal but exhibit moderate decreases in circulating T4 (25%) and T3 (13%) levels and perhaps more importantly, also exhibit extremely rapid rates of T3 entry into and escape from target tissues, presumably because there is no CRYM to sequester cytoplasmic hormone from export mechanisms. Human patients with a natural CRYM mutation that blocks its ability to bind T3 (K314T) are deaf, a known consequence of defective thyroid hormone signaling during development. Interestingly, CRYM requires NADPH for dimerization and T3 binding (Fig. 5-3). As NADPH levels are reflective of levels of cellular reducing power and anabolic capacity, it is conceivable that CRYM may sequester or release T3 in response to alterations in cellular metabolic status. This notion is attractive but not proven.
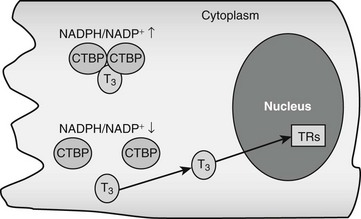
FIGURE 5-3 Thyroid hormone interactions with CTBP. Intracellular T3 can bind CTBP dimers and become sequestered in the cytoplasm. CTBP dimer formation is sensitive to intracellular NADPH/NADP+ concentrations such that free hormone will probably be released in response to reductions of NADPH levels.
There could be other T3-binding proteins in addition to CRYM and the nuclear TRs (which are described later). Radioisotope-labeled T3 interacts with proteins in the endoplasmic reticulum, mitochondrion, and nuclear envelope.2 These putative T3-binding proteins have not been identified, and the functional significance of these interactions is unknown.
Deiodinases in Thyroid Hormone Activation and Inactivation
The thyroid gland produces mostly thyroxine. Limited amounts of T3 and rT3 are secreted by the gland, but more than 80% of both these forms of thyroid hormone are produced in the periphery via actions of specific deiodinases, D1, D2, and D3.15,16 D1 and D2 remove the iodine group from the 5′ position of the outer thyronine ring. As T3 binds to TRs with higher affinity than T4, the actions of D1 and D2 deiodinases serve to increase thyroid hormone activity through generation of T3, and this represents a step-up in activity. Conversely, the action of D3 decreases hormone activity and represents a step down by removing the iodine group from the 5 position of the inner thyronine ring and converting T4 and T3 to rT3 and 3,3′-T2, respectively, neither of which interacts substantially with nuclear TRs at physiologic concentrations. Changes in deiodinase expression and activity are important control points in thyroid hormone signaling; they influence the production and availability of the biologically active form of thyroid hormone, T3.
D3 is important for clearance of plasma T3.17 It is highly expressed in human placenta, where it is thought to be important for protection of the developing fetus from effects of maternal thyroid hormones. D3 is also highly expressed in the brain and skin. Increased D3 levels may account for reductions of circulating thyroid hormone levels that are often observed in critically ill patients and states of inflammation. Finally, D3 is often overexpressed in vascular tumors, and this in turn can result in marked reductions in circulating thyroid hormone levels.
Alternate Metabolic Fates of Thyroid Hormone: Inactive and Active Derivatives
In addition to 5′ deiodination, T4 undergoes other modifications which are generally believed to inactivate the hormone.18 About 45% of circulating T4 is converted to rT3 and 35% to T3. T3 and rT3 undergo further deiodinations to create T4 derivatives with all possible combinations of iodinated and deiodinated inner and outer rings (3,3′-T2, 3,5-T2, 3′,5-T2, 3′-T1, 3-T1 and T0), none of which bind significantly to nuclear TRs. T4 and T3 can also be inactivated by glucuronidation in liver, followed by secretion into the bile or by sulfation in liver and kidney and excretion in the urine.
1. Deamination of the thyroid hormone alanine side group produces acetic acid derivatives of T4 (tetraiodoacetic acid, Tetrac) and T3 (triiodoacetic acid, Triac).19 Both are exclusive products of extrathyroidal metabolism; they are undetectable in the thyroid gland. Although they comprise about 2% of total circulating thyroid hormone, they are present at high levels in liver, where Triac represents about 14% of total thyroid hormones. Tetrac and Triac are excreted in the urine, suggesting that deamination may promote thyroid hormone clearance from the body. However, Triac binds to TRs with high affinity and acts as a potent activator. It also exhibits TR isoform preference (it preferentially binds the beta form of TR) and interacts weakly with CTBP/CRYM, suggesting that it may evade cytoplasmic sequestration by this protein. While these observations raise the issue that Triac has a distinct function from T3, this idea is not proven.
2. Decarboxylation of thyroid hormone alanine side group produces amine derivatives (thyronamines) that are directly analogous to different iodinated forms of thyroid hormone.10 Thyronamines have been detected in the thyroid gland, central nervous system, fat, and other tissues. They are bound to transporter proteins in the circulation and, though completely quantitative analyses of circulating thyronamine levels have not been published, early estimates of their abundance suggest that they are present at concentrations that are at least similar to or greater than rT3 or T3.10 At the time of writing, an endocrine role for thyronamines has not been formally proven, but there is clear evidence of biological activity. Each thyronamine has been chemically synthesized and tested in cell culture in animal models; one derivative with a single iodine substituent at the 3 position of the inner thyronine ring (T1-amine) binds and activates a cell surface G-coupled protein receptor (trace amine–associated receptor 1; TAAR1) at physiologic concentrations (subnanomolar). T1-amine induces profound hypothermia, bradycardia, and reduced cardiac output in mice and alters metabolism in several species; it reduces metabolic rate and increases lipid utilization in a pattern that is reminiscent of sleep or hibernation. T1-amine may prove to be an important endocrine hormone, and it is interesting to suggest that T4 derivatives such as rT3 may be metabolic precursors of this compound.
Actions of Thyroid Hormone
Thyroid hormones exert profound influences on nearly all tissues (Fig. 5-4).2 Receptors for thyroid hormones (TRs) are expressed fairly ubiquitously, although well-defined target tissues such as liver and heart express higher levels of TRs than others, and there are differences in distributions of particular TR isoforms20 (described in more detail in a following discussion). Essentially, thyroid hormone is needed for proper growth and development of the fetus and children and exerts widespread influences on multiple aspects of metabolism in adults.
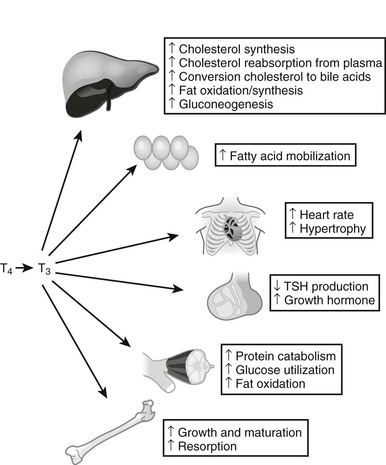
FIGURE 5-4 Tissue effects of thyroid hormones: summary of thyroid hormone effects on different tissues.
Analysis of gene expression patterns in mouse knockout models has confirmed that TRs mediate nearly all thyroid hormone responses, and that many of these effects involve changes in gene expression. While actual influences of thyroid hormone on gene expression are complex, there are important underlying principles.2 First, developmental defects that arise in cretinism and children born to hypothyroid mothers are not reversed by later hormone replacement, whereas adults with thyroid hormone imbalances exhibit metabolic disturbances that in most cases can be readily reversed by restoring TH levels to correct levels. This implies that hormone must trigger key developmental events in defined temporal windows, but thyroid hormone–regulated genes involved in metabolic regulation remain sensitive to alterations in hormone levels and continuously couple gene expression to thyroid status. Consideration of gene expression patterns, described in more detail later, reveals that some genes are induced in multiple tissues (D1 is an example), whereas other target genes are regulated in a manner that is highly tissue- and context-specific. Thus, thyroid hormone is a primary regulator of some genes, but hormone must cooperate with other factors to induce expression of other genes. Specific effects of thyroid hormone follow.
Basal Metabolic Rate
One of the most important effects of thyroid hormone involves changes in basal metabolic rate (BMR) in multiple tissues.21–24 Thyroid hormones stimulate oxygen consumption (indicative of enhanced metabolism) in multiple locations, including skeletal muscle, liver, kidney, and intestine. Increases in BMR are probably partly related to increased mitochondrial activity and number. Importantly, thyroid hormones induce expression of uncoupling proteins (UCPs), mitochondrial membrane proteins that dissipate the proton gradient in the absence of ATP synthesis, thereby converting potential energy to heat. This effect is probably a contributor to thyroid hormone–dependent increases in BMR.
Tissue Effects
Liver
Thyroid hormones exert multiple effects on the liver.2,25 They are potent mitogens in this tissue, especially in growing animals. Thyroid hormone also influences multiple metabolic processes. There is stimulation of fatty acid β-oxidation and gluconeogenesis, both key aspects of fasting response.26 However, thyroid hormone can also stimulate expression of enzymes involved in lipogenesis and generation of NADPH-reducing equivalents that are required for fat synthesis and protection against reactive oxygen species. Since it is thought that fat oxidation and synthesis do not occur simultaneously, these processes are probably separated spatially or temporally, and thyroid hormone must cooperate with other signaling mechanisms to regulate these effects.
Muscle2
Thyroid hormones promote muscle catabolism and increase skeletal muscle energy expenditure in adults. Thyroid hormone induces the insulin-sensitive glucose transporter in muscle and also promotes fat burning.30 These effects may help to sensitize muscle to insulin response. Accordingly, a human D2 gene polymorphism that reduces activity of the enzyme in muscle leads to a 20% reduction in insulin-dependent glucose disposal in humans and is correlated with insulin resistance in human populations that harbor this alteration.
Intestine
Thyroid hormone is required for normal maturation of the small intestine. TRα1 is required for proliferation of intestinal epithelial progenitor cells via induction of the β-catenin proto-oncogene.34
Systematic Analysis of Thyroid Hormone Target Genes
Many thyroid hormone–responsive genes involved in the responses described earlier are known.2 For example, in liver, thyroid hormone induces carnitine palmitoyl transferase 1a (that mediates the rate-limiting step in fatty acid oxidation); glucose-6-phosphatase and phosphoenolpyruvate carboxykinase (rate-limiting steps in gluconeogenesis); the NR coregulator PGC-1α, which is an important transcriptional coregulator that stimulates genes important for mitochondrial biogenesis, gluconeogenesis, and fat oxidation; CYP-7a1 (cholesterol-to–bile acid conversion); fatty acid synthetase; acetyl-CoA carboxylase; malic enzyme (increases fat synthesis); and spot 14, which is required for fat synthesis in some tissues and up-regulated in contexts in which carbohydrates are converted to fat. Many other hormone-responsive genes are probably still unknown.
The earliest systematic attempt to define thyroid hormone response was performed in the late 1970s. Use of two-dimensional (2D) gels to analyze extracts from radiolabeled methionine–pulsed cells revealed that about 1% of 1000 rat liver proteins change in response to altered thyroid hormone.36,37 There were inductions and repressions, and the studies began to define the “domain” of the thyroid hormone response. Recent advances in gene-profiling technology have permitted much more detailed descriptions of hormone-dependent alterations in gene profile. Presently there have been relatively few systematic genome-wide descriptions of thyroid hormone responses.25,26,38,39 An early study of thyroid hormone action in mouse liver revealed that about 1% of mouse genes change in response to altered thyroid hormone (hypothyroid versus hyperthyroid),25 confirming estimates from 2D gels. This study revealed target genes involved in carbohydrate, fat, and amino acid metabolism and unexpected thyroid hormone–regulated pathways; for example, genes involved in apoptosis are induced. The experiment also provided insights into patterns of thyroid hormone–responsive gene expression. Most regulated genes (>65%) in liver were repressed by thyroid hormone. This preponderance of negatively regulated genes is not seen in all tissues. Target genes are generally up-regulated by hormones in human muscle primary culture. Later studies of thyroid hormone patterns of gene expression in TR knockout mice provided insights into TR-specific effects and subtle differences in thyroid hormone–responsive gene expression and are discussed in more detail later in the chapter. Systematic screening of thyroid hormone–responsive genes should provide detailed descriptions of novel target genes and pathways in different tissues.
Thyroid Hormone–Regulated Micrornas
MicroRNAs (miRs) are small (18 to 25 nucleotides), noncoding RNAs that hybridize with coding mRNAs to inhibit translation and, in some cases, also promote mRNA degradation.40 The enormous roles these miRs play in regulation is just beginning to be appreciated. MiRs are produced by cleavage and processing of large primary transcripts, which often code for proteins. Thus, many of the influences that regulate expression of parental primary transcripts also regulate expression of the associated miR. While effects of thyroid hormone on miR expression have not been studied extensively, one miR (miR-208) lies within a noncoding region of the thyroid hormone–responsive human αMHC transcript (and is therefore up-regulated by thyroid hormone itself). MiR-208 is implicated in up-regulation of stress-dependent cardiac responses. It is likely that other thyroid hormone–responsive miRs exist, and they may play important roles in hormone effects on protein translation.
History of the Mechanism of Action of Thyroid Hormones
As the physiologic effects of thyroid hormones became known, investigators focused on roles of these hormones on stimulating the basal metabolic rate and on the liver.1,2 It was originally proposed that the hormones directly uncouple oxidative phosphorylation and that the primary site of action of thyroid hormones was at the mitochondria; here, investigators related actions of thyroid hormones to those of dinitrophenol. Later studies revealed other influences of thyroid hormones, including cholesterol reduction, that were hard to relate to mitochondrial actions. The notion that thyroid hormones directly uncouple mitochondrial oxidative phosphorylation did not provide a coherent unifying hypothesis to explain all actions of thyroid hormone.
In the late 1960s, Tata and colleagues conducted a number of measurements of the effects of thyroid hormones on the liver.41 He found that thyroid hormone–dependent increases in metabolic rate were blocked by actinomycin D, an inhibitor of RNA and, secondarily, protein synthesis. Also, there was an increased uptake of radiolabeled uridine into trichloric acid (TCA) precipitable material following administration of TH to animals, as well as increased activity of RNA polymerase, suggesting that TH stimulates RNA synthesis. Similar studies were being conducted with NR ligands such as with glucocorticoids, estrogens, and progestins, and there was great controversy as to whether these ligands acted through (1) transcriptional control, (2) precursor uptake, or (3) posttranscriptional control. The experiments did not address high specificity of TH responses, and as a result, controversy remained as to which of the three mechanisms applied to thyroid hormone action. Nevertheless, this study represented the first fundamentally correct proposal for the mechanism of thyroid hormone action.
Discovery of Thyroid Hormone Receptors and Their Mechanism of Action
TRs were identified in the 1970s.42,43 Following the discovery of several nuclear receptors, including the estrogen and glucocorticoid receptors, investigators in the thyroid hormone field utilized similar approaches to look for TRs. Specific binding of radiolabeled T3 to liver cells was initially discovered by Oppenheimer and colleagues using intact cells and later characterized further by Samuels and colleagues, who found that the mechanism of TR actions differed mechanistically from the steroid receptors, which translocate into the nucleus on ligand binding. For TRs, hormone could bind directly to nuclei, and the location of receptors did not alter with T3.
In the late 1970s and early 1980s, multiple aspects of the receptors were revealed.36,44 Analysis of properties of TRs revealed a single class of high-affinity binding sites with Kd values for T3 in the 0.1 nM range. Specific hormone-binding sites were observed in extracts of several responsive tissues, including liver, anterior pituitary, brain, and heart. Estimates of the number of binding sites per cell suggested that the proteins are rare; highly responsive tissues only contained about 10,000 specific hormone-binding sites per cell. Evidence that the receptor preparations correspond to physiologic hormone targets came from observations that affinities for different thyroid hormones parallel their biological potencies (Triac>T3>T4>rT3). Partial TR purification and photo-affinity labeling revealed two major nuclear hormone-binding proteins with molecular weights of 46 and 57 kD, which were later found to correspond to products of distinct genes—the TR α and β isoforms—described later in the chapter. TRs were also found to be associated with chromatin, and preferentially with active chromatin, suggesting that they influence gene expression through interactions with DNA.45–48






